

The Enigma of Amyloid Forming Proteins: Insights From Molecular Simulations*
Nevena Todorova A and Irene Yarovsky A B
A and Irene Yarovsky A B
A School of Engineering, RMIT University, Melbourne, Vic. 3001, Australia.
B Corresponding author. Email: irene.yarovsky@rmit.edu.au
Australian Journal of Chemistry 72(8) 574-584 https://doi.org/10.1071/CH19059
Submitted: 5 February 2019 Accepted: 6 March 2019 Published: 9 April 2019
Abstract
Molecular level insight into the interplay between protein sequence, structure, and conformational dynamics is crucial for the comprehensive understanding of protein folding, misfolding, and aggregation phenomena that are pertinent to the formation of amyloid fibrils implicated in several degenerative diseases. Computational modelling provides insight into protein behaviour at spatial and temporal resolution still largely outside the reach of experiments. Herein we present an account of our theoretical modelling research conducted in collaboration with several experimental groups where we explored the effects of local environment on the structure and aggregation propensity of several types of amyloidogenic peptides and proteins, including apolipoprotein C-II, insulin, amylin, and amyloid-β using a variety of computational approaches.
Introduction
The transformation of normally soluble peptides and proteins into intractable amyloid deposits has been associated with over 50 debilitating diseases, including Alzheimer’s, Parkinson’s, and type-II diabetes. Amyloid diseases have a high global presence and are a significant socioeconomic burden that poses major challenges for public health in the 21st century. This has resulted in an explosion of research to better understand the structures and mechanisms of protein aggregation and amyloid formation in an effort to develop novel, specific, and highly effective drugs for amyloid disorders.[1,2] In particular, a detailed knowledge of reactive species structure at different stages of amyloid formation is essential to optimise the design of therapeutic compounds.
Over 36 proteins, with little structural or sequence similarities, are known to form disease-related amyloid fibrils.[2] While these fibrils share a common cross β-sheet architecture, their supramolecular morphologies vary widely and can range from small protofilaments to extensive cable or rope-like structures.[3] The toxic species in amyloid diseases are generally believed to be the small, intermediate oligomers rather than the full-grown fibrils.[2] Recently this view has been expanded by a study which showed that only a small but critical concentration of amyloid fibrils are needed to catalyse the formation of toxic oligomeric species from monomeric peptide molecules.[4] There are several excellent reviews that describe the vigorous and innovative research investigating the pathogenicity and different therapeutic approaches to amyloid formation.[1,5–7] A possible therapeutic strategy against amyloid diseases could be the application of specific mutant or truncated variants that prevent the nucleation of the fibrils or assist in their dissociation. Another approach is the development of small molecules capable of preventing amyloid fibrillogenesis through different interfering mechanisms,[8] such as native-structure stabilisers, monomer sequesters, protein-aggregation inhibitors, peptide mimics, and molecular chaperones. A major impediment to the development of effective anti-aggregation compounds is their inability to cross the blood–brain barrier. A suggested solution to this predicament is the utilisation of bio-inert nanoparticles as efficient therapeutic agents or drug delivery vehicles.[9] However, aggregation processes are multiplex and protein specific, therefore it is crucial to have a molecular-level understanding of the protein structure and behaviour at different stages of amyloid formation in order to design compounds that will not only inhibit the development or induce destruction of oligomers and fibrils (treatment), but also hamper the formation of fibrils that catalyse the nucleation of the toxic oligomers (prevention).
While advances in experimental techniques are constantly probing ever-smaller length-scales and ever-shorter timescales, computational modelling is still widely recognised as an invaluable and complementary approach to systematically investigate the detailed mechanisms of nanoscale biological phenomena at atomistic and electronic resolutions.[10–12] Despite the exponential increase in computer power[13] there is no single molecular modelling approach, with the electronic structure calculations and classical all-atom and coarse-grained molecular dynamics (MD) simulations commonly used separately or in combination to describe complex biomolecular events at different length and timescales. Indeed, a multiscale modelling approach is required to obtain a comprehensive physicochemical description of multistage protein aggregation processes and, further, to design fibril inhibiting compounds and/or external stimuli. At the same time, all-atom biomolecular simulations remain the most commonly used technique to study the amyloid protein structure at the level of individual molecules, intermediate aggregates, and pre-formed fibrils.[14–16] This is because all-atom MD simulations have proven invaluable in elucidating the conformational space and dynamics of peptides and proteins, membrane transport, and ligand binding phenomena, to name a few.[17–20] In the context of this review, for example, we recently used atomistic simulations to clarify how small molecules (photoacids) bind to an all-atom model of the insulin fibril surface in order to identify a possible new means to track fibril formation by insulin.[21] More recently, we used a combination of electronic structure calculations and classical MD simulations to elucidate the mechanisms by which metal–phenolic networks are able to inhibit fibril formation by the Alzheimer’s disease implicated amyloid-β protein, which enabled us to shed light on this recently discovered phenomenon.[22]
It must be noted, however, that despite the consistent growth of modelling applications and some evident success in employing simulations to understand the elusive process of amyloid fibril formation, there are still methodological challenges that must be carefully considered. Specifically, in-silico emulated biomolecular behaviour is governed by the accuracy of the underlying potential energy function, or forcefield, which describes all inter-atomic interactions in classical molecular mechanics simulations.[23–25] Special challenges are related to identifying the existing forcefields suitable for the simulation of inherently disordered proteins, many of which have the amyloid fibril forming capacity. Hence, we and others have systematically evaluated forcefields for several proteins such as insulin[24] and human amylin[23,26] and reported significant biases of some forcefields commonly used for biomolecular simulations, which can be especially ‘influential’ when modelling inherently disordered proteins. Furthermore, the adequate sampling of the protein’s conformational space not only depends on the forcefield, it also relies on the ability of the employed approach to explore the potential energy landscape in order to have a realistic representation of the molecular transitions during various stages of the amyloid fibril formation process. Therefore, sampling methods are commonly tested alongside the choice of forcefield in an attempt to reproduce scarcely available experimental data or to test the simulation settings against one another.[23,24,27,28] In recent years we have applied several emerging sampling methods, such as bias-exchange metadynamics and replica exchange methods, to evaluate the extent to which they can access the conformational space of amyloid forming proteins, including insulin[29] and amylin.[23]
The main focus of the present review, however, is on our efforts to better understand the effects of local environment on the structure, dynamics, and self-assembly mechanisms of the amyloidogenic apolipoprotein C-II (apoC-II) and its peptide derivatives. Apolipoproteins belong to a family of proteins that are known to readily form amyloid fibrils, including apolipoprotein (apo) A-I, apoA-II, apoA-IV, apoC-II, apoC-III, apoE, serum amyloid A, and α-synuclein.[30,31] ApoC-II is a 79 amino acid protein with an important physiological role as activator of lipoprotein lipase and is a well characterised member of the apolipoprotein group, both experimentally and computationally. Its amphipathic nature allows it to circulate in the bloodstream while bound to very-low-density lipoproteins. Lipid-free apoC-II, as well as several other apolipoproteins, readily aggregate in vitro to form distinct fibrils with all the hallmarks of amyloid.[32] Amyloid fibrils of apoA-I, apoA-II, and apoC-II have been found in atherosclerotic lesions, co-localised with the amyloid marker, serum amyloid P, and have been associated with the progression of cardiovascular disease.[33] A recent in vivo study demonstrated apoC-II as the main protein component in a new type of hereditary renal amyloidosis.[34] Furthermore, an epidemiological study showed apoC-II is associated with cardiovascular disease, stroke, and myocardial infarction.†[35]
Lipid-Free ApoC-II Amyloid Fibril: Structure and Dynamics
In lipid-depleted conditions, apoC-II readily self-assembles into homogeneous fibrils with a ‘twisted-ribbon’ morphology that exhibits all the features of amyloid, including increased β-structure character, red–green birefringence in the presence of Congo Red, and increased fluorescence in the presence of thioflavin T.[32] Several experimental characterisation techniques, including fluorescence resonance energy transfer (FRET), atomic force microscopy (AFM), X-ray diffraction, and hydrogen/deuterium exchange were complemented by molecular dynamics (MD) simulations to propose a first all-atom structural model for the apoC-II amyloid fibril with a three-layered, ‘letter-G-like’, β-strand–loop–β-strand arrangement.[36] Our fully solvated all-atom MD simulations showed that the model contained a stable cross-β-core with a flexible connecting loop devoid of persistent secondary structure (Fig. 1). The simulation trajectories revealed that the charged residue clusters in the fibril rearrange to minimise the effects of same-charge interactions inherent in parallel in-register models. Our structural model for apoC-II fibrils suggested that apoC-II monomers fold and self-assemble to form a stable cross-β-scaffold containing relatively unstructured connecting loops. In a later study, our MD simulations confirmed that the core fibrillar region residues 56–76 favoured a parallel cross-β arrangement.[37]
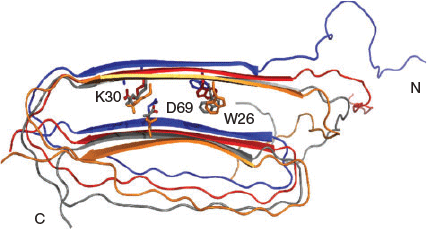
|
One important feature of this ‘letter-G-like’ model is the presence of a buried ion-pair between residues K30 and D69, as depicted in Fig. 1. These residues are present in a lipid binding region and the highly conserved α-helical lipoprotein lipase-activating region, respectively. The demonstrated formation of the buried K30–D69 charge pair within apoC-II fibrils reconciled the observed dual abilities of apoC-II to form a class A amphipathic helix and cross-β structure in lipid and lipid-free environments, respectively. Mutation studies of these residues indicated the ion-pair plays a significant role in the fibril formation process.[37–39] In our combined experimental and computational study we described the effects of D69K mutation on the structural properties of full-length apoC-II amyloid fibrils.[39] The MD simulations indicated reduced β-strand content for a model D69K apoC-II tetramer compared with the wild-type (WT) tetramer and confirmed an expansion of the cross-β spacing that contributed to the formation of a stable charge pair between K69 and E27 (Fig. 2). The computational results corroborated the experimental data that indicated more rapid fibril formation and increased β-sheet spacing in D69K apoC-II fibrils compared with the WT fibrils.

|
In a follow up study, we showed that the buried charged residues form both intra- and inter-subunit ion-pair interactions that stabilise the fibril.[38] Mutations of the ion-pair to K30D reduced the fibril stability and prevented fibril formation by K30D apoC-II under standard conditions. In contrast, co-incubation of K30D apoC-II with other mutants, i.e. mixtures of K30D apoC-II with wild-type, D69K, or double mutant (K30D/D69K) apoC-II, promoted the incorporation of K30D apoC-II into hybrid fibrils with increased stability. The MD simulations showed that an increase in the number of inter-subunit ion-pair interactions accompanied the increased stability of the hybrid fibrils. These results demonstrate the important role of both intra- and inter-subunit electrostatic interactions in stabilising apoC-II amyloid fibrils, a process that may be one of the key factors in determining the general ability of proteins to form amyloid fibrils.
ApoC-II Peptide Derivatives: Structure and Dynamics
ApoC-II contains two well protected regions between residues 19–37 and 57–74, identified by hydrogen/deuterium exchange and NMR experiments, which contain the sequences believed to be facilitating the fibril formation of apoC-II.[40] It has been shown that the synthetic apoC-II(56–76) readily formed fibrils, albeit with a different morphology compared to the analogous full-length apoC-II.[41] Further truncating the synthetic peptides narrowed this fibril-forming region to residues 60–70 and these residues were postulated to be the key driver behind fibril formation by apoC-II. This discovery culminated in several computational studies on monomeric and oligomeric apoC-II that complemented the experimental efforts of our collaborators. Below we describe over a decade of our theoretical–experimental exploration of the structure, dynamics, and fibril-forming propensity of these amyloidogenic peptides under various conditions, including variation of pH,[42] mutations,[41–43] varying lipid concentration,[42,44] cyclic peptide isomer,[45,46] presence of nanoparticles,[47] and more recently external electromagnetic fields.[48]
Mutation
Our mutation studies of the apoC-II peptide derivates show the important role of individual residues in fibril formation kinetics, dynamics, and stability.[41–43] For example, while wild-type apoC-II(56–76) and (56–76)Met60Gln peptides both readily assembled into fibrils with similar fibrillation rates, methionine oxidation resulted in slower fibril formation kinetics and a less flexible structure; however, fibril formation was not completely inhibited. Mutation of Met60 to Val, (56–76)Met60Val, caused an even slower fibril formation kinetics and a tendency of the mutant to explore a wider conformational space. Interestingly, this mutation exhibited totally different aggregation kinetics compared with the full-length apoC-II with the same mutation. Our simulation studies showed that the behaviour of apoC-II(56–76) is different to that of the full length apoC-II and this suggests two things: that the mechanism for fibril formation may likewise be different, and that smaller peptide derivatives do not necessarily serve as predictive models for fibril formation of the full length proteins.[41]
In addition, our experimental partners have shown that when the smaller apoC-II(60–70) peptides are synthesised with the same Met60 mutation, they also exhibit different fibrillation kinetics to that of the longer apoC-II(56–76) variant, as determined by thioflavin T fluorescence.[43] The oxi-Met and Met60Gln apoC-II(60–70) peptides displayed a lower rate of fibrillation, while the wild-type and Met60Val peptides formed fibrils faster. To help understand these observations, our MD simulations revealed that all apoC-II(60–70) peptides adopted conformations resembling a β-hairpin structure, albeit with some notable differences. The formation of a strong hydrophobic core, as exhibited in a β-hairpin structure, was identified to be favourable for inter-peptide interactions and subsequent fibril formation. The oxidised and Met60Gln apoC-II(60–70) monomers displayed increased structural flexibility and a broad range of aromatic side-chain orientations, while the fibril-forming wild-type and Met60Val peptides exhibited more entropically restricted β-hairpin conformations with the aromatic side-chains positioned on the opposite faces of the hairpin structure (Fig. 3). These differences were suggested to explain the distinct fibrillation kinetics observed for these mutated variants experimentally, indicating that the aromatic residues play a critical role in the fibrillation mechanism of the apoC-II(60–70) peptide.[43]

|
pH and Lipids
The fibrillation kinetics of the shorter apoC-II(60–70) peptide was investigated at different pH and in the presence of lipids.[42,44,49] Our MD simulations revealed distinct preferences in apoC-II(60–70) secondary structure pertaining to the change in pH and the presence of lipids, thus identifying specific fibril-favouring or fibril-disrupting conditions. The conformational differences were then compared with the peptide behaviour at the same experimental conditions. At fibril-favouring conditions (neutral and low pH) the peptide preferentially adopted β-hairpin structures, with the aromatic residues Tyr63 and Phe67 lying on the opposite faces of the hairpin, while under fibril-disruptive conditions (lipid-rich) significantly different conformations were preferred by the peptide, with the aromatic residues Tyr63 and Phe67 orientations at the same face of the hairpin.
Lipid concentration has been shown to strongly influence the structure and aggregation propensity of apoC-II(60–70) peptide.[44] The MD results showed a progressive reduction in the solvent accessible surface area of apoC-II(60–70) with increasing lipid concentration, accompanied by increased lipid–peptide contacts. The peptide exhibited reduced conformational flexibility due to the persistent lipid–peptide interactions. A significant change in the secondary structure of apoC-II(60–70) peptide was also observed with increasing lipid concentration. At lower concentrations (1–3 lipids per peptide), the peptide adopted extended β-strand conformations, caused by contacts with the lipids which reduced the intramolecular interactions within the peptide. In contrast, a higher lipid concentration (4–6 lipids per peptide) had a restraining effect on the peptide’s flexibility by trapping it in a particular conformation. Such behaviour was deemed to be inhibiting the fibril formation, because of the lipid-induced inability of the peptide to adopt fibril competent conformations. This observation concurred with the experimental finding which revealed that the 4 : 1 lipid-to-peptide ratio is sufficient to cause fibril inhibition in apoC-II(60–70).
Cyclic Peptides
Cyclic peptides and their derivatives have been receiving growing attention as potentially powerful and highly specific fibril inhibitors ([50] and references therein), but also as molecules that are capable of decreasing the cytotoxicity of fibril aggregates.[51] What makes cyclic peptides so appealing is their resistance to enzymatic degradation, i.e. they are slow to metabolise compared with their non-cyclic equivalents. However, cyclic peptides are prone to aggregation due to hydrophobic interactions and this can reduce their fibril inhibitory efficacy. An excellent review discusses the latest strategies for designing cyclic peptides, specifically the importance of peptide amino sequence and/or conformational similarity to amyloid fibrils.[50]
To explore if cyclicity could be exploited to inhibit apoC-II fibril formation, we investigated if a cyclic apoC-II(60–70) derivate, cyc(60–70), reduced the fibrillogenic nature of its linear-peptide analogue when in a co-mixture.[45,46] Cyc(60–70) was formed by disulfide cross-linking of cysteine residues added to the termini of the linear apoC-II(60–70). This cyclic peptide did not self-associate into fibrils, however, substoichiometric concentrations of cyc(60–70) significantly delayed fibril formation by the fibrillogenic linear peptides apoC-II(60–70) and apoC-II(56–76).[45] Reduction of the disulfide bond or scrambling the amino acid sequence within cyc(60–70) significantly impaired its inhibitory activity, suggesting that the mechanism of aggregation inhibition is sequence specific. The solution structure of cyc(60–70) was solved using NMR spectroscopy, revealing a well defined amphipathic structure. MD simulations were employed to refine the structure of cyc(60–70) and compare to its scrambled variants. The simulations identified a flexible central region within cyc(60–70), while the ‘scrambled’ cyc(60–70) exhibited an increased formation of intramolecular hydrogen bonds and a reduction in the overall flexibility of the peptide. Our structural studies suggested that the inhibitory activity of cyc(60–70) is mediated by an elongated structure with inherent flexibility and distinct amphipathic (hydrophobic and hydrophilic) surfaces, enabling cyc(60–70) to interact transiently with fibrillogenic peptides and inhibit fibril assembly.
Understanding the mechanisms by which these cyclic molecules block the formation of amyloid fibrils can help design specific therapeutic agents to prevent amyloidosis. With this in mind, we employed a MD-based umbrella sampling approach together with electronic structure calculations to elucidate the mechanisms of inhibition and affinity between cyc(60–70) and apoC-II(60–70) peptide.[46] Our results showed that cyc(60–70) induced increased flexibility in apoC-II(60–70), suggesting that cyc(60–70) inhibits fibrillation of the linear analogue by destabilising the apoC-II(60–70) structure, rendering it incapable of adopting fibril competent conformations. In contrast, cyc(60–70) itself became less flexible upon binding to apoC-II(60–70), which was mediated by hydrophobic interactions between the aromatic rings of the peptides. This cyclic–linear peptide complex effectively created a ‘cap’ around the fibril-forming region of apoC-II(60–70) and generated an outer hydrophilic shell that discouraged further apoC-II(60–70) self-assembly. Our potential of mean force simulations in solution together with under vacuum DFT calculations of the cyclic–linear peptide binary complex showed that apoC-II(60–70) has a stronger binding affinity for the hydrophobic face of cyc(60–70) compared with the hydrophilic side (Fig. 4). This suggested the amphipathic ‘Janus’-like character of cyc(60–70) to be an important feature of potential molecular candidates for fibril inhibition, a property to be exploited in the design of specific inhibitors of amyloid fibril formation.

|
Electromagnetic Fields
The upsurge of electronic ‘smart’ devices has caused human exposure to electromagnetic fields (EMF) to be inescapable. This has resulted in an increased concern about potential health effects of this omnipresent exposure. We have been contributing to research in this area through our previous modelling of insulin exposure to various electric fields[52–55] and more recently, through our involvement into the Australian Center of Electromagnetic Bioeffects Research (ACEBR).[56] High-level fields of specific frequency can excite certain vibrational modes of proteins and other biomolecules, causing structural changes that can lead to misfolding and ultimately, aggregation of proteins into insoluble amyloid fibrils.[52–54,57,58] On the other hand, experimental studies have suggested that EMFs may be used as a therapeutic tool for the breakdown of fibrils implicated in Alzheimer’s disease.[59] Therefore, an improved understanding of the effects of radiofrequency radiation on biomolecules and their implications in disease processes at the molecular level is required. Such knowledge will not only assist in setting reliable safety standards for proliferating mobile electronic devices but will also help clarify whether EMF exposure can have any positive health related effects, specifically in certain neurodegenerative processes. We have chosen to investigate the effects of radiofrequency radiation on the amyloidogenic apoC-II(60–70) peptide, well studied by us and others both theoretically and experimentally, thus providing ample benchmarking opportunities for assessing any potential EMF effects on its behaviour.
To identify and systematically characterise molecular-level effects of EMF on the behaviour of apoC-II(60–70) peptide we performed explicit solvent MD simulations where we varied the electric field strength from a commonly used high strength field of 0.7 V nm−1 [52,58] to a relatively low intensity field of 0.0007 V nm−1.[48] This approach enabled us identify the lowest field strength at which we can reproducibly detect and characterise the field effects on the structure and dynamics of the peptide model and relate the changes to the fibril forming capacity of the peptide identified through our previous work.[42–44,47] Our simulations showed field strengths lower than 0.004 V nm−1 had no tangible effects on the peptide conformation. ApoC-II(60–70) adopted amyloid-prone hairpin structures similar to those in ambient conditions (Fig. 5b). The intermediate field-strength range (0.04–0.004 V nm−1) caused a significant increase in peptide dynamics, which resulted in an increased population of structures with fibril-inhibiting characteristics, such as the separated N- and C-termini and colocation of the aromatic residues at the same peptide face. In the high strength field (> 0.04 V nm−1) simulations, apoC-II(60–70) experienced peptide dipole alignment along the applied field direction, which resulted in elongated structures and disrupted the inherent β-hairpin conformation known to be the intermediate state for fibril formation (Fig. 5a). These findings suggested that intermediate-strength electromagnetic fields could be considered for designing alternative treatments of amyloid diseases, while the very high and low field strengths could be employed for engineering well ordered fibrillar aggregates (e.g. biocompatible materials) for other medicinal or non-medicinal applications. We are currently investigating the effects of high frequency EMF in the 1.0–10 GHz range (suggested for the new mobile telecommunication technologies, e.g. 5G network[60]) on the structure and dynamics of the apoC-II-derived peptide at the lowest field intensity level at which the effects were theoretically measurable.
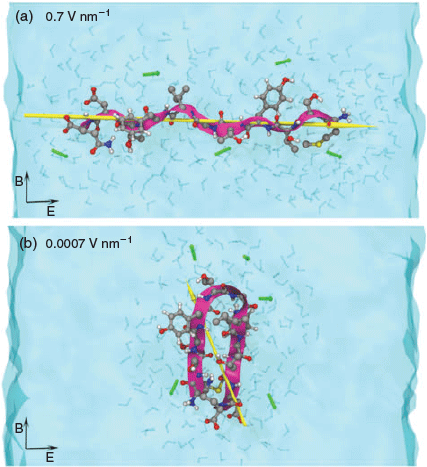
|
Nanomaterials
The proliferation of nanotechnology in every industry has made nanomaterials ubiquitous in the natural world, including plants, animals, humans, and the environment. While some nanomaterials have been shown to be beneficial or functional in nanomedicine and technology, others can be detrimental to our health and the environment, with many having unforeseen and unintended consequences.[61,62] Operating in the nano-world, these materials require a nanoscale description of their interactions with the surrounding molecules to ensure their programmability and safety.[63] Several reviews summarise the plethora of research describing the common modes in which nanostructured materials interact with biological molecules such as membranes, DNA, and peptides/proteins, and, within our current focus, their possible role in protein aggregation.[64–68] Despite the extensive number of studies in this field there is still a lot to learn about nanomaterial interactions with biological matter. Specifically in relation to amyloid formation, nanomaterials may inhibit or promote cytotoxicity at three different stages of the fibrillation process: (1) by disruption of the nucleation phase, i.e. the formation of fibril seeds, (2) by influencing the elongation/fibril growth ability, and (3) by altering the amount of toxic species present and or forming.[69] Nanomaterials of varying composition, functionalisation, shape, and size have been shown to affect the fibrillation of amyloidogenic peptides and proteins in many different ways. Factors such as shape, size, and surface chemistry, including concentration and composition of surface functionalisation (and charge), have been shown to impact the ability of nanoparticles to inhibit or promote aggregation. Considerable research has been undertaken recently to explore how these factors specifically affect fibril formation ([64–67] and references therein).
Graphitic Nanomaterials
One of the most studied and prevalent types of nanomaterials present in the environment are carbonaceous nanomaterials. Through the process of combustion, they are continually ejected in large volumes into the atmosphere as airborne particles. In the laboratory they can be synthesised at the smallest scale, in the form of clusters or flakes with nanometric dimensions and amazing mechanical, thermal, and optical properties. Graphitic nanomaterials are characterised by trivalent carbon atoms with the sp2 hybridisation placed in a two-dimensional lattice. While this lattice can adopt many structures, arguably the three most commonly known nano-carbon morphologies are spherical fullerenes, tubular carbon nanotubes, and flat graphite and graphene surfaces. These unique structures give graphitic nanoparticles distinct properties which are desirable in applications ranging from electronics, catalysis, chemical sensing, biosensors, drug and vaccine delivery, and many more. Fullerenes, particularly C60, can neutralise free radicals in solution. This makes them an attractive therapeutic for the treatment of Alzheimer’s disease as oxidative stress has been implicated in the pathogenesis of this terrible disease.[70] Carbon nanotubes (CNTs) have been considered for many uses in vivo including as biosensors, as site-specific drug delivery vehicles, or for direct interaction with disease-related biomolecules involved in amyloidosis.[71] However, it remains unclear how these largely hydrophobic nanomaterials can potentially affect any undue and undesirable amyloid formation due to non-specific peptide/protein adsorption and aggregation in proximity of hydrophobic surfaces.[72,73] To clarify this we again used our well studied apoC-II(60–70) as a prototype peptide for benchmarking the effects of 0D, 1D, and 2D carbon nanomaterials on its capacity to aggregate.
Classical MD and electronic structure calculations (linear scaling DFT)[74] were employed to investigate the effects of carbonaceous nanoparticles of differing size and curvature on the structure, dynamics, and binding affinity of the amyloidogenic apoC-II(60–70) peptide.[47] Our results showed that the binding affinity of apoC-II(60–70) peptide to C60, nanotubes, and graphene decreases with increasing nanoparticle curvature (Fig. 6). Strong binding to graphene and nanotubes was facilitated by the large contact area available for π-stacking between the aromatic residues of the peptide and the extended graphitic surfaces. The highly curved fullerene surface exhibited reduced efficiency for π-stacking, however it contributed to an increase in conformational lability of apoC-II(60–70), which prevented it from adopting fibril-favouring structural features, such as a stable β-hairpin. This finding is in line with the previous studies of oxidised apoC-II(60–70), where increased structural flexibility and dynamics were shown to be the key factors prohibiting this peptide to form fibrils, confirmed experimentally.[42,43] In contrast, the nanotube and graphene surfaces promoted extended, entropically restricted fibril forming peptide conformations.

|
Advanced sampling MD simulations were also applied to study the effects of graphitic carbon nanomaterials on the structure, and the dissociation pathway of a previously identified preformed dimer of the amyloidogenic apoC-II(60–70) peptide.[75] Similarly to our monomeric peptide study, the calculated free energy of dissociation showed that the apoC-II(60–70) dimer weakly bound to C60 nanoparticles but strongly interacted with elongated carbon nanotubes and graphene. The significant curvature of the C60 surface contributed to the weaker peptide complex binding to the nanoparticle due to an increase in the peptide dynamics, which resulted in dissociation of the dimer from the C60 surface. In contrast, stronger interactions were observed between the elongated carbon nanomaterials and the apoC-II(60–70) dimer and this resulted in a separation of the dimer complex with one monomer strand remaining adsorbed on the surface of the nanomaterial during an in-silico pull-off experiment. This demonstrated that the interaction between the bound peptide and flat graphitic surfaces is stronger than the inter-peptide interactions between the peptide strands within the dimer complex. The study suggested that flat surface carbon nanomaterials present favourable binding substrates for aromatic-rich peptides, and thus have the ability to act as templates to mediate peptide self-assembly and fibril growth, while the highly curved C60 particle can act as an inhibitor of fibril formation.
Like graphene, its allotropes have also attracted much attention in the past several years, with the most intriguing allotrope being graphene oxide (GO). Unlike its counter-parts, GO is not constrained by complicated production techniques and its synthesis procedures have been well established.[76] GO has the chemical formula of C10O1(OH)1(COOH)0.5,[77] which can be visualised as two epoxy groups, two hydroxy groups on both sides of the basal plane, and one carboxy group on the edge for every 20 carbon atoms. Due to its oxygen containing functionalised surface, GO sheets show excellent hydrophilicity, are relatively easy to disperse in water, and can be easily transported and handled, relative to plain graphene sheets. GO has a large and highly functionalisable surface which allows for non-covalent interactions with biological molecules via electrostatics, hydrogen bonding, and π–π stacking. Several experimental studies identified GO nanoparticles as potential inhibitors of fibril formation.[78,79] Despite some computational efforts,[80,81] the mechanisms of interactions of GO with biological milieu is not well understood. The lack of consistent parameters for modelling GO nanoparticles and the variation in oxidation of GO has commanded the need for improved characterisation of GO nanomaterials. Recently, we investigated how varying the size and oxidation of GO flakes can affect their structural and dynamic properties in an aqueous solution.[82] Our all-atom modelling of the GO nanoflakes of different sizes suggested that the curvature and roughness of relatively small (3 × 3 nm) GO flakes were not affected by their degree of oxidation, while the larger (7 × 7 nm) flakes exhibited an increase in surface roughness as their oxidation increased. The degree of oxidation induced a well structured first hydration layer, which manifested in identifiable hydrophobic and hydrophilic patches on GO. Our simulations provided improved models and understanding of GO nanoflake structure, which can be used in future as a guide for the rational design of functional graphitic nanoparticles for modulation of the amyloid formation.
Gold Nanomaterials
Owing to the unique size-dependent chemical, physical, and optical properties and relative ease in functionalisation, gold nanoparticles (AuNPs) are showing vast potential for application in several biomedical industries (see Yeh et al.[83] and references therein). The implications of AuNPs in fibril formation in particular have been described at length in a recent review.[68] There is also a growing research interest in understanding the complex physicochemical phenomena occurring at the Au–bio interface, and the following perspective outlines the current successes and challenges associated with the multiscale computational treatment of Au–bio interfacial systems.[12]
We recently applied computer simulations to investigate the facet-dependent conformational changes of another amyloidogenic protein, human amylin, on AuNPs, and their role in fibril formation.[84] Human amylin (IAPP) is responsible for glycemic regulation in our body, however, like the other amyloid proteins, it can self-assemble and form fibrils which are involved in the pathogenesis of type II diabetes.[85] We have recently explored the conformational preferences of IAPP in solution[23] which provided some background for studying the effects of AuNP on its conformation and surface induced aggregation. The AuNPs used for this study experimentally were 2, 5, and 10 nm in diameter. To emulate the surface of the larger nanoparticles, we modelled the most featured facets present on gold, the Au(111) and Au(100) facet individually. Each facet was capped with citrate molecules at the experimentally determined concentration and immersed in physiological solution composed of explicit solvent and salt. The mechanisms of interactions of monomeric and fibrillar amylin on the citrate-coated Au(111) and Au(100) surface were investigated using all-atom MD simulations starting from different orientations to exclude any interaction bias. The combined experimental and computational simulation study demonstrated a strong interaction between full-length IAPP and AuNPs, which was initiated by the hydrophilic N-terminal domain, followed by a conformational change dependent on the Au surface facet. The IAPP–AuNP interaction was observed to accelerate IAPP fibrillation by the preferential formation of ordered structures, which was attributed to the AuNP-induced formation of an intermediate helix and conformational restructuring of the amyloidogenic region that consequently promoted IAPP fibrillation. Our MD simulations showed significant conformational rearrangements on Au(111) facets, the dominant crystallographic facet in larger AuNPs (Fig. 7). Adsorption of IAPP to gold was primarily driven by electrostatic interactions with the citrate adlayer. The role of water in the mechanisms of binding of IAPP to the Au surfaces was also investigated, with the simulations showing a higher density of water on the Au(100) surface (water-mediated binding) compared with the Au(111) surface. This effect was due to the square arrangement of Au atoms on Au(100) surfaces, which facilitated denser structuring of water, and marks a distinct difference to the in-plane quasi-hexagonal atomic arrangement of the Au(111) surface. The demonstrated ability of AuNPs to modulate interactions with the IAPP enabled us to suggest a new way of preventing membrane disruption by the IAPP using gold nanomaterials.[84]

|
Summary and Conclusions
We have presented an overview of our theoretical modelling studies on protein structure and dynamics implicated in self-assembly and amyloid fibril formation. The effect of various ‘stresses’ including residue specific oxidation or substitution (mutation), and external factors such as pH, lipids, cyclic peptide, electromagnetic fields, and different nanoparticles on the protein conformation and fibril-forming ability were explored in atomic detail using coarse grained, atomistic, and electronic structure calculation methods.
Our studies showed that theoretical simulations are a valuable tool for structure refinement and, more importantly, for identifying specific structural features in protein/peptide models that are present under fibril-forming or fibril-inhibiting conditions, which is currently undetectable by conventional experiments. Some of the structural characteristics of the apoC-II(60–70) peptide present under fibril-forming conditions include the formation of a hydrophobic core formed by a β-hairpin conformation and aromatic side-chain arrangement on the opposite face of the peptide. Whereas under fibril-inhibiting conditions, increased conformational dynamics and aromatic side-chains on the same side of the amyloidogenic peptide were identified. These structural features can be typical for various amyloidogenic peptides and can serve as potential targets for the design of fibril inhibiting mutations or compounds. As our mutation studies demonstrated, single residue substitutions can have profound effects on protein structure and dynamics and can promote or inhibit fibril formation depending on the mutant’s chemistry and (co)-location. We have also demonstrated that cyclic peptide derivatives of amyloidogenic linear peptides can be designed as specific inhibitors of amyloid fibril formation. Furthermore, low intensity electromagnetic fields can be employed to destabilise and potentially destroy amyloid fibrils, which can lead to new treatments of amyloid-related diseases. At the same time, electromagnetic fields of sufficiently high intensity can be used to stimulate protein denaturation and field related alignment to produce novel biocompatible fibrous materials, which can be exploited in biomedical and industrial applications.
Our simulations also showed that nanoparticles can act as fibril inducers or inhibitors with the outcome ultimately depending on their size, curvature, and chemistry. Similarly to the amyloid mediation cases summarised above, graphitic and gold nanoparticles can engage the same interaction mechanisms to prevent protein self-association in solution by targeting the structural features that drive self-assembly. The aromatic regions of graphitic nanoparticles enable π–π stacking with amyloidogenic proteins, disrupting a crucial mechanism that leads to fibril aggregation through self-association and aromatic stacking. Gold nanoparticles, on the other hand, have demonstrated fibril promoting effects by acting as a nucleation centre for fibril growth. This mechanism effectively reduced the amount of free proteins in solution, which in-turn, decreased the number of toxic fibrillar species formed, as shown experimentally. Overall, it is clear that a comprehensive and systematic understanding of the mechanisms of interaction between biomolecules and nanomaterials is crucial for the development of novel diagnostic and therapeutic approaches.
From a computational perspective, it is evident that brute-force simulations alone are largely insufficient due to their limitation to access the time and length scales of biological phenomena such as ligand binding and self-assembly. This calls for enhanced sampling techniques such as umbrella sampling, bias-exchange metadynamics (BE-META), or replica exchange with solute-tempering (REST), capable of exploring the slow and ‘rare’ events inaccessible by continuous MD simulations. Furthermore, a multiscale approach, which combines quantum mechanical, classical, and coarse-grained methodologies, is currently the best option for a wholesome and comprehensive description of complex biological phenomena. In conclusion, in-silico studies prove to be a pivotal complementary approach to experiments in the quest for a more complete understanding of the complex behaviour of proteins, where theoretical modelling can guide the design and development of therapies for protein conformation related diseases.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
Collaborating teams of A/Prof. Geoff Howlett (Bio21, the University of Melbourne), Dr Herbert Treutlein (SANOOSA Pty Ltd), and Prof. Molly Stevens (Imperial College London, UK) are gratefully acknowledged for providing inspiration and guidance (and invaluable experimental data!) for this research. The contribution of RMIT postgraduate students and postdoctoral researchers to the research presented is acknowledged, including that of Dr Andrew Hung, Dr Sue Legge, Mr Levi Yeung, Mr Enxi Peng, Mr Alan Bentvelzen, Dr Adam Makarucha, and Dr Akin Budi. Special thanks to Dr Patrick Charchar for useful discussion and proof-reading the manuscript. IY thanks the Australian Research Council (ARC) for providing funding for several projects described herein (LP0219406; LP0562041; DP0984565; DP140101888; DP170100511) and the National Health and Medical Research Council (NHMRC) for providing funding for the Australian Center of Excellence for Electromagnetic Bioeffects Research (grants CRE1042464 and CRE1135076). The research projects were undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI, project e87), which is supported by the Australian Government.
References
[1] A. Aguzzi, T. O’Connor, Nat. Rev. Drug Discov. 2010, 9, 237.| Crossref | GoogleScholarGoogle Scholar | 20190788PubMed |
[2] T. P. Knowles, M. Vendruscolo, C. M. Dobson, Nat. Rev. Mol. Cell Biol. 2014, 15, 384.
| Crossref | GoogleScholarGoogle Scholar | 24854788PubMed |
[3] R. Nelson, D. Eisenberg, Curr. Opin. Struct. Biol. 2006, 16, 260.
| Crossref | GoogleScholarGoogle Scholar | 16563741PubMed |
[4] S. I. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson, T. P. Knowles, Proc. Natl. Acad. Sci. USA 2013, 110, 9758.
| Crossref | GoogleScholarGoogle Scholar | 23703910PubMed |
[5] A. J. Doig, P. Derreumaux, Curr. Opin. Struct. Biol. 2015, 30, 50.
| Crossref | GoogleScholarGoogle Scholar | 25559306PubMed |
[6] M. Bartolini, V. Andrisano, ChemBioChem 2010, 11, 1018.
| Crossref | GoogleScholarGoogle Scholar | 20401887PubMed |
[7] V. Kumar, N. Sami, T. Kashav, A. Islam, F. Ahmad, M. I. Hassan, Eur. J. Med. Chem. 2016, 124, 1105.
| Crossref | GoogleScholarGoogle Scholar | 27486076PubMed |
[8] T. Hard, C. Lendel, J. Mol. Biol. 2012, 421, 441.
| Crossref | GoogleScholarGoogle Scholar | 22244855PubMed |
[9] S. I. Yoo, M. Yang, J. R. Brender, V. Subramanian, K. Sun, N. E. Joo, S.-H. Jeong, A. Ramamoorthy, N. A. Kotov, Angew. Chem. 2011, 50, 5110.
| Crossref | GoogleScholarGoogle Scholar |
[10] S. Bottaro, K. Lindorff-Larsen, Science 2018, 361, 355.
| Crossref | GoogleScholarGoogle Scholar | 30049874PubMed |
[11] D. J. Huggins, P. C. Biggin, M. A. Dämgen, J. W. Essex, S. A. Harris, R. H. Henchman, S. Khalid, A. Kuzmanic, C. A. Laughton, J. Michel, A. J. Mulholland, E. Rosta, M. S. P. Sansom, M. W. van der Kamp, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, e1393.
| Crossref | GoogleScholarGoogle Scholar |
[12] P. Charchar, A. J. Christofferson, N. Todorova, I. Yarovsky, Small 2016, 12, 2395.
| Crossref | GoogleScholarGoogle Scholar | 27007031PubMed |
[13] P. J. Denning, T. G. Lewis, Commun. ACM 2017, 60, 54.
| Crossref | GoogleScholarGoogle Scholar |
[14] N. V. Buchete, R. Tycko, G. Hummer, J. Mol. Biol. 2005, 353, 804.
| Crossref | GoogleScholarGoogle Scholar | 16213524PubMed |
[15] L. Tran, T. Ha-Duong, Peptides 2015, 69, 86.
| Crossref | GoogleScholarGoogle Scholar | 25908410PubMed |
[16] I. M. Ilie, D. Nayar, W. K. den Otter, N. F. A. van der Vegt, W. J. Briels, J. Chem. Theory Comput. 2018, 14, 3298.
| Crossref | GoogleScholarGoogle Scholar | 29715424PubMed |
[17] R. O. Dror, R. M. Dirks, J. P. Grossman, H. Xu, D. E. Shaw, Annu. Rev. Biophys. 2012, 41, 429.
| Crossref | GoogleScholarGoogle Scholar | 22577825PubMed |
[18] M. Karplus, R. Lavery, Isr. J. Chem. 2014, 54, 1042.
| Crossref | GoogleScholarGoogle Scholar |
[19] D. E. Shaw, P. Maragakis, K. Lindorff-Larsen, S. Piana, R. O. Dror, M. P. Eastwood, J. A. Bank, J. M. Jumper, J. K. Salmon, Y. Shan, W. Wriggers, Science 2010, 330, 341.
| Crossref | GoogleScholarGoogle Scholar | 20947758PubMed |
[20] S. J. Marrink, V. Corradi, P. C. T. Souza, H. I. Ingolfsson, D. P. Tieleman, M. S. P. Sansom, Chem. Rev. 2019, in press.
| Crossref | GoogleScholarGoogle Scholar | 30758191PubMed |
[21] N. Amdursky, M. H. Rashid, M. M. Stevens, I. Yarovsky, Sci. Rep. 2017, 7, 6245.
| Crossref | GoogleScholarGoogle Scholar | 28740173PubMed |
[22] W. Zhang, A. J. Christofferson, Q. A. Besford, J. J. Richardson, J. Guo, Y. Ju, K. Kempe, I. Yarovsky, F. Caruso, Nanoscale 2019, 11, 1921.
| Crossref | GoogleScholarGoogle Scholar | 30644497PubMed |
[23] E. Peng, N. Todorova, I. Yarovsky, PLoS One 2017, 12, e0186219.
| Crossref | GoogleScholarGoogle Scholar | 29023509PubMed |
[24] N. Todorova, F. S. Legge, H. Treutlein, I. Yarovsky, J. Phys. Chem. B 2008, 112, 11137.
| Crossref | GoogleScholarGoogle Scholar | 18698702PubMed |
[25] V. H. Man, X. He, P. Derreumaux, B. Ji, X. S. Xie, P. H. Nguyen, J. Wang, J. Chem. Theory Comput. 2019, 15, 1440.
| Crossref | GoogleScholarGoogle Scholar | 30633867PubMed |
[26] K. Q. Hoffmann, M. McGovern, C. C. Chiu, J. J. de Pablo, PLoS One 2015, 10, e0134091.
| Crossref | GoogleScholarGoogle Scholar | 26221949PubMed |
[27] M. Carballo-Pacheco, A. E. Ismail, B. Strodel, J. Chem. Theory Comput. 2018, 14, 6063.
| Crossref | GoogleScholarGoogle Scholar | 30336669PubMed |
[28] G. H. Zerze, C. M. Miller, D. Granata, J. Mittal, J. Chem. Theory Comput. 2015, 11, 2776.
| Crossref | GoogleScholarGoogle Scholar | 26575570PubMed |
[29] N. Todorova, F. Marinelli, S. Piana, I. Yarovsky, J. Phys. Chem. B 2009, 113, 3556.
| Crossref | GoogleScholarGoogle Scholar | 19243106PubMed |
[30] C. L. Teoh, M. D. W. Griffin, G. J. Howlett, Protein Cell 2011, 2, 116.
| Crossref | GoogleScholarGoogle Scholar | 21400045PubMed |
[31] G. J. Howlett, T. M. Ryan, M. D. W. Griffin, Biochim. Biophys. Acta, Proteins Proteomics 2018, in press.
[32] D. M. Hatters, C. E. MacPhee, L. J. Lawrence, W. H. Sawyer, G. J. Howlett, Biochemistry 2000, 39, 8276.
| Crossref | GoogleScholarGoogle Scholar | 10889036PubMed |
[33] C. R. Stewart, A. Haw, R. Lopez, T. O. McDonald, J. M. Callaghan, M. J. McConville, K. J. Moore, G. J. Howlett, K. D. O’Brien, J. Lipid Res. 2007, 48, 2162.
| Crossref | GoogleScholarGoogle Scholar | 17630380PubMed |
[34] S. H. Nasr, S. Dasari, L. Hasadsri, J. D. Theis, J. A. Vrana, M. A. Gertz, P. Muppa, M. T. Zimmermann, K. L. Grogg, A. Dispenzieri, S. Sethi, W. E. Highsmith, G. Merlini, N. Leung, P. J. Kurtin, J. Am. Soc. Nephrol. 2017, 28, 439.
| Crossref | GoogleScholarGoogle Scholar | 27297947PubMed |
[35] R. Pechlaner, S. Tsimikas, X. Yin, P. Willeit, F. Baig, P. Santer, F. Oberhollenzer, G. Egger, J. L. Witztum, V. J. Alexander, J. Willeit, S. Kiechl, M. Mayr, J. Am. Coll. Cardiol. 2017, 69, 789.
| Crossref | GoogleScholarGoogle Scholar | 28209220PubMed |
[36] C. L. Teoh, C. L. L. Pham, N. Todorova, A. Hung, C. N. Lincoln, E. Lees, Y. H. Lam, K. J. Binger, N. H. Thomson, S. E. Radford, T. A. Smith, S. A. Müller, A. Engel, M. D. W. Griffin, I. Yarovsky, P. R. Gooley, G. J. Howlett, J. Mol. Biol. 2011, 405, 1246.
| Crossref | GoogleScholarGoogle Scholar | 21146539PubMed |
[37] C. O. Zlatic, Y. Mao, N. Todorova, Y. F. Mok, G. J. Howlett, I. Yarovsky, P. R. Gooley, M. D. W. Griffin, FEBS J. 2018, 285, 2799.
| Crossref | GoogleScholarGoogle Scholar | 29791776PubMed |
[38] N. Todorova, C. O. Zlatic, Y. Mao, I. Yarovsky, G. J. Howlett, P. R. Gooley, M. D. W. Griffin, Biochemistry 2017, 56, 1757.
| Crossref | GoogleScholarGoogle Scholar | 28229588PubMed |
[39] Y. Mao, C. O. Zlatic, M. D. W. Griffin, G. J. Howlett, N. Todorova, I. Yarovsky, P. R. Gooley, Biochemistry 2015, 54, 4805.
| Crossref | GoogleScholarGoogle Scholar | 26196342PubMed |
[40] L. M. Wilson, Y. F. Mok, K. J. Binger, M. D. W. Griffin, H. D. T. Mertens, F. Lin, J. D. Wade, P. R. Gooley, G. J. Howlett, J. Mol. Biol. 2007, 366, 1639.
| Crossref | GoogleScholarGoogle Scholar | 17217959PubMed |
[41] F. S. Legge, K. J. Binger, M. D. Griffin, G. J. Howlett, D. Scanlon, H. Treutlein, I. Yarovsky, J. Phys. Chem. B 2009, 113, 14006.
| Crossref | GoogleScholarGoogle Scholar | 19780547PubMed |
[42] A. Hung, M. D. Griffin, G. J. Howlett, I. Yarovsky, Eur. Biophys. J. 2008, 38, 99.
| Crossref | GoogleScholarGoogle Scholar | 18769912PubMed |
[43] N. Todorova, A. Hung, S. M. Maaser, M. D. W. Griffin, J. Karas, G. J. Howlett, I. Yarovsky, Phys. Chem. Chem. Phys. 2010, 12, 14762.
| Crossref | GoogleScholarGoogle Scholar | 20938536PubMed |
[44] N. Todorova, A. Hung, I. Yarovsky, J. Phys. Chem. B 2010, 114, 7974.
| Crossref | GoogleScholarGoogle Scholar | 20496878PubMed |
[45] M. D. W. Griffin, L. Yeung, A. Hung, N. Todorova, Y.-F. Mok, J. A. Karas, P. R. Gooley, I. Yarovsky, G. J. Howlett, J. Mol. Biol. 2012, 416, 642.
| Crossref | GoogleScholarGoogle Scholar |
[46] N. Todorova, L. Yeung, A. Hung, I. Yarovsky, PLoS One 2013, 8, e57437.
| Crossref | GoogleScholarGoogle Scholar | 23437387PubMed |
[47] N. Todorova, A. J. Makarucha, N. D. M. Hine, A. A. Mostofi, I. Yarovsky, PLOS Comput. Biol. 2013, 9, e1003360.
| Crossref | GoogleScholarGoogle Scholar | 24339760PubMed |
[48] N. Todorova, A. Bentvelzen, N. J. English, I. Yarovsky, J. Chem. Phys. 2016, 144, 085101.
| Crossref | GoogleScholarGoogle Scholar | 26931725PubMed |
[49] A. Hung, I. Yarovsky, J. Mol. Graph. Model. 2011, 29, 597.
| Crossref | GoogleScholarGoogle Scholar | 21146432PubMed |
[50] J. Luo, J. P. Abrahams, Chem. – Eur. J. 2014, 20, 2410.
| Crossref | GoogleScholarGoogle Scholar | 24478167PubMed |
[51] J. A. Kritzer, S. Hamamichi, J. M. McCaffery, S. Santagata, T. A. Naumann, K. A. Caldwell, G. A. Caldwell, S. Lindquist, Nat. Chem. Biol. 2009, 5, 655.
| Crossref | GoogleScholarGoogle Scholar | 19597508PubMed |
[52] A. Budi, F. S. Legge, H. Treutlein, I. Yarovsky, J. Phys. Chem. B 2005, 109, 22641.
| Crossref | GoogleScholarGoogle Scholar | 16853947PubMed |
[53] A. Budi, F. S. Legge, H. Treutlein, I. Yarovsky, J. Phys. Chem. B 2007, 111, 5748.
| Crossref | GoogleScholarGoogle Scholar | 17472363PubMed |
[54] A. Budi, F. S. Legge, H. Treutlein, I. Yarovsky, J. Phys. Chem. B 2008, 112, 7916.
| Crossref | GoogleScholarGoogle Scholar | 18537286PubMed |
[55] A. Budi, S. Legge, H. Treutlein, I. Yarovsky, Eur. Biophys. J. 2004, 33, 121.
| Crossref | GoogleScholarGoogle Scholar | 14574523PubMed |
[56] S. P. Loughran, M. S. Al Hossain, A. Bentvelzen, M. Elwood, J. Finnie, J. Horvat, S. Iskra, E. P. Ivanova, J. Manavis, C. K. Mudiyanselage, A. Lajevardipour, B. Martinac, R. McIntosh, R. McKenzie, M. Mustapic, Y. Nakayama, E. Pirogova, M. H. Rashid, N. A. Taylor, N. Todorova, P. M. Wiedemann, R. Vink, A. Wood, I. Yarovsky, R. J. Croft, Int. J. Environ. Res. Public Health 2016, 13, 967.
| Crossref | GoogleScholarGoogle Scholar |
[57] F. Mancinelli, M. Caraglia, A. Abbruzzese, G. d’Ambrosio, R. Massa, E. Bismuto, J. Cell Biol. 2004, 93, 188.
[58] N. J. English, C. J. Waldron, Phys. Chem. Chem. Phys. 2015, 17, 12407.
| Crossref | GoogleScholarGoogle Scholar | 25903011PubMed |
[59] G. W. Arendash, J. Sanchez-Ramos, T. Mori, M. Mamcarz, X. Lin, M. Runfeldt, L. Wang, G. Zhang, V. Sava, J. Tan, C. Cao, J. Alzheimers Dis. 2010, 19, 191.
| Crossref | GoogleScholarGoogle Scholar | 20061638PubMed |
[60] https://www.telstra.com.au/consumer-advice/eme (accessed 1 March 2019)
[61] X. Yang, M. X. Yang, B. Pang, M. Vara, Y. N. Xia, Chem. Rev. 2015, 115, 10410.
| Crossref | GoogleScholarGoogle Scholar | 26293344PubMed |
[62] A. Elsaesser, C. V. Howard, Adv. Drug Deliv. Rev. 2012, 64, 129.
| Crossref | GoogleScholarGoogle Scholar | 21925220PubMed |
[63] Q. X. Mu, G. B. Jiang, L. X. Chen, H. Y. Zhou, D. Fourches, A. Tropsha, B. Yan, Chem. Rev. 2014, 114, 7740.
| Crossref | GoogleScholarGoogle Scholar |
[64] M. Zhang, X. Mao, Y. Yu, C.-X. Wang, Y.-L. Yang, C. Wang, Adv. Mater. 2013, 25, 3780.
| Crossref | GoogleScholarGoogle Scholar | 23722464PubMed |
[65] B. Wang, E. H. Pilkington, Y. Sun, T. P. Davis, P. C. Ke, F. Ding, Environ. Sci. Nano 2017, 4, 1772.
| Crossref | GoogleScholarGoogle Scholar | 29230295PubMed |
[66] F. De Leo, A. Magistrato, D. Bonifazi, Chem. Soc. Rev. 2015, 44, 6916.
| Crossref | GoogleScholarGoogle Scholar | 26139348PubMed |
[67] A. J. Makarucha, N. Todorova, I. Yarovsky, Eur. Biophys. J. 2011, 40, 103.
| Crossref | GoogleScholarGoogle Scholar | 21153635PubMed |
[68] T. John, A. Gladytz, C. Kubeil, L. L. Martin, H. J. Risselada, B. Abel, Nanoscale 2018, 10, 20894.
| Crossref | GoogleScholarGoogle Scholar | 30225490PubMed |
[69] C. Li, R. Mezzenga, Nanoscale 2013, 5, 6207.
| Crossref | GoogleScholarGoogle Scholar | 23744243PubMed |
[70] E. E. Tuppo, L. J. Forman, J. Am. Osteopath. Assoc. 2001, 101, 11S.
[71] X. Xue, L.-R. Wang, Y. Sato, Y. Jiang, M. Berg, D.-S. Yang, R. A. Nixon, X.-J. Liang, Nano Lett. 2014, 14, 5110.
| Crossref | GoogleScholarGoogle Scholar | 25115676PubMed |
[72] S. Linse, C. Cabaleiro-Lago, W.-F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford, K. A. Dawson, Proc. Natl. Acad. Sci. USA 2007, 104, 8691.
| Crossref | GoogleScholarGoogle Scholar | 17485668PubMed |
[73] K. Bhattacharya, S. P. Mukherjee, A. Gallud, S. C. Burkert, S. Bistarelli, S. Bellucci, M. Bottini, A. Star, B. Fadeel, Nanomedicine (Lond.) 2016, 12, 333.
| Crossref | GoogleScholarGoogle Scholar |
[74] N. D. M. Hine, P. D. Haynes, A. A. Mostofi, C. K. Skylaris, M. C. Payne, Comput. Phys. Commun. 2009, 180, 1041.
| Crossref | GoogleScholarGoogle Scholar |
[75] A. J. Makarucha, N. Todorova, I. Yarovsky (eds.), Effects of Graphitic Nanomaterials on the Dissociation Pathway of Amyloidogenic Peptide Dimer. International Conference on Nanoscience and Nanotechnology (ICONN), 2014, 2–6 February.
[76] W. S. Hummers, R. E. Offeman, J. Am. Chem. Soc. 1958, 80, 1339.
| Crossref | GoogleScholarGoogle Scholar |
[77] G. Shao, Y. Lu, F. Wu, C. Yang, F. Zeng, Q. Wu, J. Mater. Sci. 2012, 47, 4400.
| Crossref | GoogleScholarGoogle Scholar |
[78] M. Mahmoudi, O. Akhavan, M. Ghavami, F. Rezaee, S. M. Ghiasi, Nanoscale 2012, 4, 7322.
| Crossref | GoogleScholarGoogle Scholar | 23079862PubMed |
[79] M. Mahmoudi, H. R. Kalhor, S. Laurent, I. Lynch, Nanoscale 2013, 5, 2570.
| Crossref | GoogleScholarGoogle Scholar | 23463168PubMed |
[80] L. Baweja, K. Balamurugan, V. Subramanian, A. Dhawan, J. Mol. Graph. Model. 2015, 61, 175.
| Crossref | GoogleScholarGoogle Scholar | 26275931PubMed |
[81] Y. Chen, Z. Chen, Y. Sun, J. Lei, G. Wei, Nanoscale 2018, 10, 8989.
| Crossref | GoogleScholarGoogle Scholar | 29725676PubMed |
[82] E. X. Peng, N. Todorova, I. Yarovsky, ACS Omega 2018, 3, 11497.
| Crossref | GoogleScholarGoogle Scholar |
[83] Y. C. Yeh, B. Creran, V. M. Rotello, Nanoscale 2012, 4, 1871.
| Crossref | GoogleScholarGoogle Scholar | 22076024PubMed |
[84] S. T. Wang, Y. Lin, N. Todorova, Y. Xu, M. Mazo, S. Rana, V. Leonardo, N. Amdursky, C. D. Spicer, B. D. Alexander, A. A. Edwards, S. J. Matthews, I. Yarovsky, M. M. Stevens, Chem. Mater. 2017, 29, 1550.
| Crossref | GoogleScholarGoogle Scholar | 28260837PubMed |
[85] E. T. Jaikaran, A. Clark, Biochim. Biophys. Acta 2001, 1537, 179.
| Crossref | GoogleScholarGoogle Scholar | 11731221PubMed |
* Irene Yarovsky is the recipient of the 2017 RACI Physical Chemistry Division Medal.
† I wish to dedicate this article and my research on amyloid fibrils to my parents, Emma Poliakova (1938–2018) and Yulii Gershenzon (1938–2007) – the physicists who inspired me to study. Sadly, they both fell victims to cardiovascular disease and stroke while fully intellectually active. IY