

Recent Trends Concerning Upconversion Nanoparticles and Near-IR Emissive Lanthanide Materials in the Context of Forensic Applications
William J. Gee A
A
A School of Physical Sciences, University of Kent, Giles Lane, Canterbury CT2 7NH, UK. Email: W.Gee@kent.ac.uk

William Gee graduated from Deakin University in 2006, obtaining a Bachelor of Forensic Science degree with First Class Honours. He then obtained his Ph.D. in inorganic chemistry from Monash University under the supervision of Professor Phil Andrews and Professor Peter Junk in 2011. William later won a 2012 Victoria Fellowship, and held post-doctoral positions at Monash University and the University of Bath before securing an ongoing academic teaching and research position at the University of Kent in 2017 as Lecturer in Chemistry and Forensic Science. |
Australian Journal of Chemistry 72(3) 164-173 https://doi.org/10.1071/CH18502
Submitted: 12 October 2018 Accepted: 6 December 2018 Published: 4 January 2019
Journal Compilation © CSIRO 2019 Open Access CC BY-NC-ND
Abstract
Upconversion nanoparticles (UCNPs) are materials that, upon absorbing multiple photons of low energy (e.g. infrared radiation), subsequently emit a single photon of higher energy, typically within the visible spectrum. The physics of these materials have been the subject of detailed investigations driven by the potential application of these materials as medical imaging devices. One largely overlooked application of UCNPs is forensic science, wherein the ability to produce visible light from infrared light sources would result in a new generation of fingerprint powders that circumvent background interference which can be encountered with visible and ultraviolet light sources. Using lower energy, infrared radiation would simultaneously improve the safety of forensic practitioners who often employ light sources in less than ideal locations. This review article covers the development of UCNPs, the use of infrared radiation to visualise fingerprints by the forensic sciences, and the potential benefits of applying UCNP materials over current approaches.
Introduction
Lanthanide (Ln) ions are uniquely suited to photoemissive applications. Photonic application of lanthanide materials may now be found in agriculture, biosensing, medicine, and material applications like counterfeit tags and various sensors for physical stimuli.[1] Near-infrared (NIR) emissive and absorptive species are particularly interesting from the perspective of medicine and biosensing. This is because NIR light has a high degree of tissue penetration and lower propensity to damage biological matter relative to higher-energy wavelengths.[2] Interest also surrounds incorporating these materials into security devices to harness wavelengths that are ordinarily invisible to the human eye, thereby increasing the difficulty of forging and replicating these items (Fig. 1).[1] Harnessing Ln luminescent materials to discourage and confound forgers was perhaps the first mainstream incidence of Ln materials influencing the criminal justice system; however, others have followed, for example, the recent application of volatile luminescent lanthanide species to visualise latent fingerprints.[3] Given that identifying and visualising evidence with luminescence is a common aspect of forensic science, further integration of these two fields is inevitable. Here, an overview of NIR-emissive Ln materials is first provided, followed by relevant examples of NIR applications in forensic science, and finally, the state-of-the-art prospects of marrying these two fields together in applying NIR-emissive Ln materials to forensic applications.
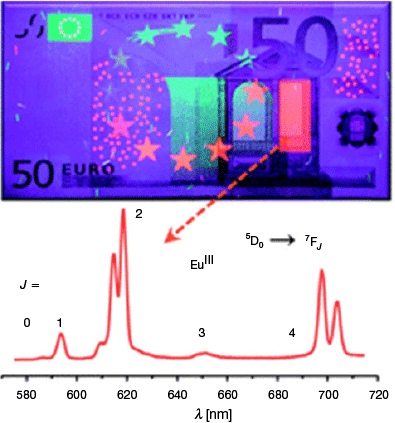
|
The predilection of Ln ions to luminesce stems from several inherent properties of these metals, one being well-defined energy transitions that result from shielding of the 4f orbitals by the 5s25p6 subshells that yield extremely narrow emission bands.[4] These emission bands are relatively unaffected by the local microenvironment (e.g. pH value or temperature),[5] provided that the lanthanide ions are adequately shielded from coordinating solvent molecules and other sources of vibrational deactivation. This latter goal can be challenging given the ubiquity of hydrogenated ligand components like C–H, O–H, and N–H moieties. Overcoming this issue typically requires a good understanding of coordination chemistry and the ability to control the Ln coordination sphere through careful choice of coordinating ligands. Once a suitable coordination environment has been established, Ln ions provide long luminescent lifetimes[6] and a broad spectrum of wavelength emissions that can be tuned from ultraviolet (UV) to NIR by simply selecting the appropriate Ln metal ion.[7]
Given most lanthanide ions are luminescent to some extent,[6] targeting NIR emission requires judicious choice of the Ln ion. Within the NIR spectral window, the ions of most interest are Pr3+, Nd3+, Ho3+, Er3+, and Yb3+.[6] Many of these ions have been investigated with the intent of applying their emissive properties across a range of applications. For example, the telecommunication window between 1.34 and 1.54 μm is perfectly suited to Nd3+ and Er3+ integrated optic materials capable of amplifying optical signals,[8] and emission at 1200 nm is suited to integration in modern high-speed local area networks (LANs).[8,9]
As alluded to above, photoemission from lanthanides in the free-ion form is highly inefficient. This is a combination of both the spin and parity forbidden nature of the required Laporte 4f–4f transitions. In simplistic terms, the solution is to transfer energy to the metal via a ligand antenna using a ligand(S1) → ligand(T1) → Ln* pathway;[6] here, the excitation of the antenna ligand results in the population of a ligand singlet state (S1) and subsequent decay via intersystem crossing to a triplet state (T1). The ligand triplet excited state is the more efficient means of energy transfer to the metal ion because it is more long-lived. This triplet state ultimately decays through either a Dexter-type or Förster-type dipole–dipole exchange mechanism to transfer energy to the Ln ion.[10–12] In reality, this process is complex and rests on an intricate balance of various rate constants that compete with nonradiative pathways. Lanthanide photoemission is thus reliant on interplay between the lowest triplet level of the Ln complex and the resonance level of the ligated Ln ion. Manipulating the triplet level can be achieved by changing the ligand, granting a measure of control over the luminescence intensity of the Ln ion.[13] To facilitate efficient energy transfer, the lowest triplet state of the antennae should lie ~3500 cm−1 above the emitting excited states of the chosen lanthanide[14]; however, in some instances a difference of 1500 cm−1 has been shown to be sufficient.[15] Given that these excited-state energies will vary depending on the identity of the Ln ion, possessing the ability to precisely tailor the energy levels provided by the ligand antennae is vital. Arguably the easiest way to achieve this is through synthetic design and manipulation of coordinating functional groups.
Ligand design further plays an important role in designing emissive Ln species given that vibrational deactivation of excited states may be triggered by C–H, O–H, and N–H moieties, which are ubiquitous to most organic species. This mode of deactivation is particularly troublesome for NIR emitters owing to their relatively low energy profile. Several strategies have been employed to combat this means of deactivation, with one common approach being to replace C–H bonds with lower-energy C–F oscillators, thereby improving NIR emission.[16,17] The challenge of controlling ligand behaviour is further complicated by the somewhat unpredictable coordination geometry of the Ln ions, coupled with a need to shield the Ln ion from coordinating solvents; however, efforts have been made to try and understand structural trends in the Ln series.[18]
Recent Developments in Ln NIR Emitters
β-Diketonate ligands have been recently shown to be adept at sensitising NIR emission from Ln metals due to their relatively low triplet energy levels.[19,20] This ligand class is capable of providing efficient energy transfer to the first excited state of many NIR-emissive lanthanides (e.g. the 4F3/2 transition for Nd3+ of 11257 cm−1 or the 2F5/2 transition for Yb3+ of 10400 cm−1), although the extent of this efficiency is influenced by ligand functionalisation.[19] This is serendipitous because diketonate ligands are well-known for their ability to strongly bind lanthanides by taking advantage of their oxophilic nature and to benefit from well-established synthetic routes to complexes and clusters.[18] Their use as NIR-radiation antennae builds upon prior work in which β-diketonates were shown to be very efficient at absorbing higher-energy light and transferring it to ligated lanthanides.[20] Indeed some diketonate variants have been shown to absorb high-energy UV radiation, thereby promoting a π–π* transition in dibenzoylmethanide ligands which, when ligated to Nd3+, give characteristic NIR emission.[21] No phosphorescence from the ligand was observed for the Tb3+ variant, which suggests efficient energy transfer; however, this is reduced for Nd3+ owing to a less favourable energy gap between the triplet excited state of the ligand and the 4F3/2 excited state of Nd3+. This understanding of the singlet and triplet energy-level gap between new β-diketonate ligands and the metal is necessary to gauge the efficiency of energy transfer to NIR-emitting Ln ions.[19] This is usually done by synthesising a gadolinium (Gd3+) complex with the new ligand, owing to the high energy of the first excited state of the gadolinium ion (5IJ 36900 cm−1), which precludes the possibility of metal-derived emission and allows a detailed study of ligand-derived emissions.[19]
Research in this area has expanded to β-triketonates in an effort to boost the NIR efficiency of Er3+ and Yb3+ complexes.[22–24] This has been effective in boosting the NIR emissive output, an outcome justified as stemming from the removal of methylene C–H oscillators in close proximity to the Ln centre.[22] Furthermore, these complexes exhibit very long luminescent lifetimes relative to that of their β-diketonate counterparts. Extended conjugated systems are inherently suited to absorbing high-energy photons, which has led to strategies of appending other, more complex chromophores (such as pyrene analogues) to further boost efficiency.[22] Challenges still remain with these current Ln-diketonate NIR materials, including low thermal stabilities, limited photostability, and poor processability, with inclusion in polymer films being actively explored as a solution to these issues.[24]
Porphyrinate ligands, which are themselves excellent light-harvesting molecules, have been functionalised with boron-dipyrromethene (BODIPY) and coupled to Yb3+ as a means of attempting efficient light harvesting and sensitising NIR emission using modular components.[25] Functionalisation of these species with various groups yielded a range of absorption profiles that range from 430 to 624 nm and multiple emission bands that cover the visible region and NIR region at 1030 nm. The higher-energy emissions were determined to originate from BODIPY, whereas the latter were derived from the Yb3+ centre. The study also observed two-photon-induced NIR emission with a maximum at 980 nm and an excitation wavelength of 800 nm, which suggests potential application as an imaging material for biological systems (see below).
Lanthanide metal–organic frameworks (MOFs) have also been touted as a modifiable scaffold that is capable of modulated NIR emission. Achieving this requires altering the functionality of the organic linkers while choosing metal nodes capable of sensitised NIR emission (Pr3+, Nd3+, Ho3+, etc.). This behaviour was exemplified by a Yb3+ MOF upon grafting of a simple amine functionality onto its organic linker, which imparted a red shift of the excitation wavelength required to produce NIR emission.[26] The measured quantum yield for this material was 1.26 %, one of the highest reported at the time. Given that this material possessed an accessible pore volume of ~24 %, opportunities exist to further perturb the emission by hosting different molecular guests within the MOF or upon guest uptake. Other examples of NIR-emissive MOFs include a Nd3+ analogue that could be sensitised using UV radiation from antenna-like organic ligands[27] and a mixed Nd3+–Yb3+ framework that makes use of a sensitiser–emitter couplet to produce various bands of NIR radiation at 890, 980, 1060, and 1350 nm upon 808 nm laser pumping.[28]
Medical Application: A Key Driver of Innovation
Improving the visualisation of cellular material is an area that is driving the development of NIR-emitting Ln species. The ideal biological transparency window extends from λ 700 to 1200 nm, which corresponds to the wavelength range that minimises photodegradation and light scattering while maximising penetration into thick tissues.[2,29,30] Furthermore, at these wavelengths, light can be more easily focussed within nanometric volumes typically probed by microscopy.[31] More attuned properties of NIR emitters to biological systems enables as good a resolution as can be achieved relative to the more emissive, visibly luminescent Ln ions (e.g. Eu3+ and Tb3+). The best NIR-emitting Ln candidates for medical applications must provide efficient two-photon emission, be both thermally and kinetically stable, and possess good photophysical properties that resist degradation. In the context of medical applications, time-gated brightness, as proposed by the groups of Maury[29] and Parker in relation to lanthanide luminescence,[32] is of key importance to optical microscopy of biological samples. Notable examples of tissue imaging that have been achieved using Ln NIR emission include a Yb3+ complex that harnessed a two-photon sensitisation method using a home-made NIR-to-NIR biphotonic microscopy set-up.[33] This work was later expanded to include Sm3+, which enabled combined visible and NIR Ln emission to be simultaneously achieved in living cells.[5,29] Refinement of the Ln coordination environment resulted in exceptional luminescent emission from NIR-emissive Ln species in water, enabling bioprobing of HeLa cells.[5,34] Similarly, 3D networks of blood capillaries in slices of mouse brain stained with a Yb complex were visualised using NIR-to-NIR biphotonic microscopy.[33] These highlights are but a small subset of the collective work being done in this area, and the reader is referred to several recent reviews for a more in-depth overview.[35–41]
Lanthanide NIR Upconversion Nanoparticles
Upconversion luminescence is an anti-Stokes process wherein a luminescent centre, here a lanthanide ion, sequentially absorbs multiple photons of lower energy to produce a single photon with higher energy (Fig. 2).[42,43] Lanthanide metal centres are particularly suited to this process because they typically possess a range of metastable excited states with relatively long lifetimes (~ms). This means that upon absorption of the initial photon, a longer window of opportunity exists in which secondary interactions with additional photons may occur. This process is distinct from two-photon absorption-based luminescence (i.e. excited state absorption, ESA, Fig. 3, left) in that it can be triggered using low-powered and incoherent light sources. Metal-ion-derived upconversion under low-power, continuous wave radiation is uniquely suited to the lanthanide ions Er3+, Ho3+, and Tm3+, given their large anti-Stokes shifts, sharp emission profile, and high photostability.
|

|
Two upconversion processes, energy transfer upconversion (ETU, Fig. 3, centre) and cooperative upconversion (CUC, Fig. 3, right) make use of sensitising Ln metals to harvest low-energy photons and transfer them to an acceptor lanthanide ion tasked with emitting the higher-energy photon.[4,44,45] Great interest surrounds these materials stemming from better prospects of applications arising from low-energy-triggered upconversion relative to their more energy-intensive two-photon absorption counterparts. Materials capable of visible-light upconversion from common light sources, including solar radiation, are highly sought after, and presently, there are a wide array of emerging applications for upconversion nanoparticles (UCNPs).[46]
The radiation emitted from these Ln-doped UCNPs can range from the UV to the NIR by taking advantage of the long lifetimes of the excited states produced by the sequential addition of multiple lower-energy photons.[47] In general, the emissive properties of lanthanide ions are highly reliant on the crystal field. For UCNP emission, lower symmetry is preferable to higher symmetry because these environments enhance the electronic coupling of the 4f energy levels and their higher electronic configurations, which increases the 4f–4f transition probabilities.[4] Placing the Ln3+ ion in a dielectric field serves to split the 4f energy levels as a consequence of the local crystalline environment of the metal, which in turn has a strong influence on emission intensity.
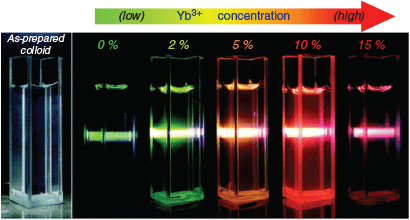
Most NIR Ln UCNP materials rely on two components: a lanthanide dopant and the dielectric host lattice it is embedded in. The dopant is typically present in a concentration <1 % and is required to sequentially absorb two (or more) low-energy photons in order to release a single higher-energy photon. This requires a match in energy between that of the incoming NIR photon and the energy band between the ground state and the excited state of the Ln metal. Once an electron has been promoted to the excited state, the dopant may interact with a second photon, aided by the long-lived metastability of the excited state. This results in the release of a photon with the combined energies of absorbed lower-energy photons in the form of upconverted luminescent emission. The lanthanide elements Er3+, Tm3+, and Ho3+ are commonly used as dopants because these ions possess energy gaps that are well-matched to the energy of the incoming NIR photons.[4,6] These ions have multiple emission bands (e.g. Er3+ at 650 and 540 nm), and hence by manipulating the population of these bands, the emitted-light wavelength can be varied. The energy level diagrams of Er3+, Tm3+, and Ho3+ are shown in Fig. 4, which highlight that NIR photons (dashed red arrows) of different wavelengths (e.g. 1490 or 808 nm) can be used to excite different metastable excited states of each ion, allowing relaxation with emission of higher-energy photons of variable wavelengths (coloured solid arrows).[48]
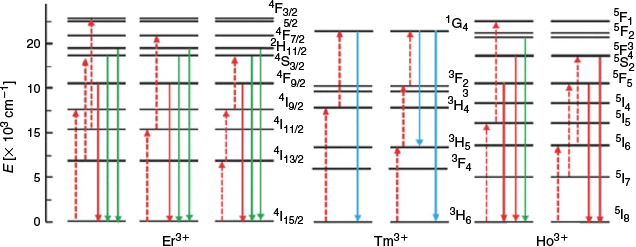
|
Pairs of lanthanide metal ions may also participate in upconversion through a process of energy transfer upconversion or cooperative upconversion. These occur when two or more neighbouring ions each absorb NIR photons and adopt metastable states. One metastable species then transfers its energy to the neighbouring ion, enabling emission of a higher-energy photon (Fig. 3). The efficiency of the luminescent output by this mechanism is low; however, this can be offset by the addition of multiple donor ions per emissive ion. However, too large an excess of donor ions can induce a quenching effect. This leads to a distinction between activators and sensitisers for dopant lanthanides. Because Yb3+ possesses a relatively simple energy level diagram (i.e. Fig. 5), it is typically used as a sensitiser given its large absorbing cross-section at ~980 nm (2F7/2–2F5/2) that matches the energy level of other activators (Er3+, Tm3+, and Ho3+).[49] Nd3+ has also been used as an alternate sensitiser paired with Er3+.[45,50,51]
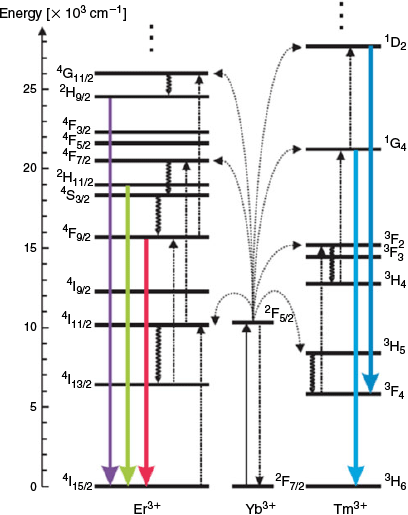
|
Considerable theoretical and experimental studies have elucidated the mechanism behind many upconversion materials doped with Ln3+ ions.[4,52,53] From this understanding, five general strategies for manipulating the emission of UCNPs have been identified: variation of the host lattice, tailoring of the crystal field around the lanthanide dopants, plasmonic enhancement, nanoscopic control of the energy transfer process, and suppression of surface-related deactivation.[45,47] The first two strategies, variation of the crystalline host and manipulation of the crystal field of the lanthanide dopants, are discussed further here. Key host properties include transparency in the spectral window of interest, resistance to optical damage, and overall structural stability. Dielectric hosts possessing these traits also require very low frequency phonons in order to inhibit nonradiative pathways leading to de-excitation.[47] The archetypical lattice is composed of simple yttrium salts, such as brightly emitting NaYF4,[54–56] or YVO4 that provides a narrower luminescence spectrum containing less red emission.[49,57] Manipulating the crystal field is arguably easy to achieve but difficult to control. Co-doping the crystalline host with other ions has been used as a strategy to improve emissive properties,[58–61] with disruption of lattice symmetry identified as a potential cause of this enhancement.[62] Manipulating the ligands contained within the host that interact with the coordination sphere of the metal is also a viable route for improved emission characteristics. Incorporation of dye molecules in one such UCNP emitter effected a 3000-fold increase in emission owing to its superior antenna effect for the Ln3+ ion.[63] Refining the identity and crystalline environment of Ln ions within new UCNPs is the heart of recent synthetic efforts in the area and has been a key driver in the discovery of new upconversion mechanisms.
Although Fig. 3 depicts two common means of generating upconversion, new mechanisms taking advantage of different combinations of lanthanides continue to emerge, exemplified by two recent examples: energy migration-mediated upconversion (EMU)[64] and the thermal avalanche mechanism.[65] Discovery of these new mechanisms of upconversion has been driven by the synthesis of new materials, in which judiciously chosen metals fill a range of roles including sensitiser, accumulator, migrator, and activator. Given the emergent nature of these materials, low quantum yields remain a barrier to commercialisation of this technology, coupled with the difficulty in evaluating and recording the upconversion emission spectra using commercial fluorescence spectrometers.[4]
Progress has been made in tuning the emission profile of UCNPs, despite difficulty in achieving high quantum yields. Strategies for achieving this have included altering the concentration of dopant, phase, size, and morphology of the material, and by varying the ligand identity.[66,67] The excitation band of UCNPs has also been broadened through design with the ultimate aim of applying this research to improve the efficiency of solar cells.[4,68] This has been achieved by focussing on embedding transition metals into the material or by integrating NIR dyes as antenna molecules to harvest light across a wider spectrum of wavelengths. One latter example involved grafting the NIR dye IR-780 into the UCNP, which resulted in an increase in efficiency across the system by 3300 times and broadened the excitation band more than 3-fold (ranging over 720–1000 nm).[63]
Discrete Ln UCNP Materials
As mentioned above, the archetypical Ln UCNP materials are Ln ions doped within a 3D lattice composed of yttrium salts. One recent interesting variation on this approach has been the development of discrete molecules that contain the minimal molecular machinery to generate upconversion. These complexes utilise the same energy transfer processes as Ln-doped UCNPs and retain both a sensitising and emitting Ln ion (typically Er3+). Early studies of dimeric complexes that comprised of a single sensitiser coupled to an acceptor ion were generally unsuccessful in generating upconversion, which was justified to be a result of very short lifetimes of the excited state.[69] In silico calculations were then made for trinuclear complexes that comprised of a single acceptor Ln ion sandwiched between two sensitiser species. This arrangement was predicted to improve the upconversion emission intensity by two orders of magnitude. Further improvements in emission intensity could be gained when a longer-lived, sensitiser-centric pathway using Cr3+ was implemented, with an expected increase in intensity of 7–8 times predicted.[69] The desirability of chromium as a sensitiser stems from the six lower excited levels of its [Ar] 3d3 electronic configuration that can be tuned using ligand-field effects.[70] Efforts to realise these predictions consequently focussed on lanthanide-chromium mixed-metal species to maximise potential improvement of emission. Evaluation of Cr-Er-Cr trinuclear clusters was performed by Piguet et al. under the reasoning that emission would be favoured owing to the alignment of the three metals along a pseudo-3-fold axis and that the chosen polydentate ligands would both shield the metals from solvent-triggered nonradiative relaxation and regulate the interatomic spacing of the Cr-Er-Cr complex.[44] These helical complexes are stabilised by polypyridine ligands and were found to emit characteristic erbium-derived (4S3/2–4I15/2) green emission upon excitation with a 750 nm laser source. Manipulation of ligand binding kinetics to inhibit dissociation of the Cr-Er-Cr core motif was further found to be essential for stimulating NIR upconversion at higher temperatures, including room temperature, as would be needed for many target applications.[71] These studies demonstrate that molecular design approaches may be implemented to tailor complexes with optimised NIR upconversion built around a central lanthanide emitter such as Er3+.
Dimeric complexes were later revisited, resulting in the identification of weak but measurable upconversion for dimeric Cr-Er complexes ligated by polypyridyl ligands. Excitation of the chromium centres harnessed 750 nm NIR radiation, which instigated a spin-flip of the (2E–4A2) and (2T1–4A2) energy transitions, that upon transference to the erbium centre resulted in green upconversion (540 nm, 4S3/2–4I15/2).[70] These dimeric species were more susceptible to dissociation than their trinuclear analogues. However, the approximate 9 Å separation between the Cr and Er centres in the complex boosts efficiency of energy transfer to almost 50 %.[44]
Dual Ln Emitting and Upconverting Materials
Very few reports exist of materials capable of producing both NIR emission as well as upconversion. One such report describes an Er3+ complex ligated by 8-hydroxyquinoline that when sublimed as a thin film yields both upconverted emission in the UV spectrum and ligand-sensitised photoluminescence.[72] The absorption spectra shows weak bands in the region of 480–580 nm, corresponding to the 4f–4f transitions of Er3+, and very strong absorption with a maximum at 380 nm originating from the ligands. Consequently, the most efficient strategy to trigger photoemission was to target the antenna ligands, which facilitate allowable transitions to the metal ion. Unlike Er3+-doped nanoparticles, multiple phonon relaxation was not the dominant mode of deactivation for the excited states. Instead, a broad photoluminescent response in the visible region was observed originating from the organic ligands. Interestingly, this emission profile extended all the way into the NIR region and could be modulated based on the wavelength of excitation. Upconversion could be triggered by the application of an argon laser with a wavelength of 514.5 nm, which resulted in at least a three-photon absorption to deliver the highest wavelengths of UV radiation emitted by the complex.
Harnessing NIR Luminescence for Fingermark Visualisation
Visualising latent fingermarks (i.e. invisible fingerprints) at crime scenes is a fundamental aspect of forensic science. This stems from the ridge patterns present on a person’s finger pads remaining unchanged during the course of a lifetime, excepting injuries that penetrate to the underlying dermis layer.[73] There are up to three levels of ridge detail ranging from generic fingerprint pattern (Level 1) to specific locations of sweat pores within ridges (Level 3) that make fingermarks entirely unique to an individual, even for monozygotic twins.[74]
Crime scene examiners commonly employ fingerprint dusting powders to develop latent fingermarks in a process that has changed remarkably little in over a hundred years. The key aim of dusting for fingerprints is to produce contrast between the developed fingermarks and the surface on which they reside. This becomes challenging for multicoloured, patterned, or variably textured surfaces.[75] The first innovation to address this issue was the development of luminescent fingerprint powders that could be made to fluoresce or phosphoresce upon stimulation, usually with UV radiation.[74,75] The luminescent emission of these powders is overwhelmingly in the visible spectrum (400–700 nm); however, problems remain for fluorescent surfaces, which are increasingly common given the prevalence of emissive dyes, polymers, and advanced materials such as polymer money.[76] NIR emission has been identified as a key means of eliminating background interference from difficult surfaces. This is because far fewer materials fluoresce when exposed to visible light or NIR radiation than for UV radiation, and because many materials that are troublesome under UV radiation readily absorb IR light, and consequently an IR emitter will produce excellent contrast.[76] Additionally, given that IR photography has a long history of use in forensic science,[77] the means of capturing this mode of contrast is readily available. NIR emission has been evaluated for existing fingermark treatments, typically those producing visible dyes or fluorescent emitters. These have included diazafluoren-9-one, 1,2-indanedione, and genipin treatments of paper, as well as cyanoacrylate/rhodamine 6G treatment of plastics.[78] Substituted styryl dyes were identified as a facile means of imparting NIR emission from a cyanoacrylate-treated piece of evidence.[79] Similarly, fingerprint powders coated in a NIR dye have been used to promote NIR emission. Both of these posttreatments could be improved upon by development of inherently NIR-emitting fingermark materials.
NIR–NIR fluorophores similarly represent extremely promising materials for forensic science, owing to their potential to suppress virtually all types of background luminescent emissions. This is because illuminating a material with visible light can promote background artefacts to appear in the NIR region, and because the security features of many advanced materials (polymer banknotes, passports, etc.) are inert in the NIR window.[80] The application of an 850 nm long-pass filter is typically sufficient to screen out all radiative emissions other than from the powder, including that of higher-energy NIR excitation wavelengths.
Recognition of this has stimulated considerable research in the area of nanoparticles, either for direct application as powders or in liquid suspensions to improve safety. Progress to this effect has been highlighted in an extensive review by Bécue.[81] These nanomaterials have encompassed heavy-metal-based quantum dots (QDs),[82–84] noble-metal nanocomposites,[85] lanthanide-based phosphors (see below),[54,57,86–91] and many others.
Current NIR Emitters for Fingermark Visualisation
Development of inherently NIR-emitting powders initially focussed on materials containing chlorophyll or anthocyanin as the active component.[76] Chlorophyll absorbs light over much of the visible spectrum and emits strongly in the NIR region. Similarly, anthocyanins absorb lower-wavelength visible light (typically >600 nm) and re-emit at lower energies, from 650 nm into the NIR region. Given that both are sourced from plant matter, suitably powdered organic material from wheatgrass, spinach, kale, lime, and others all show NIR emission when excited by 640 nm light.[76] The source of organic material was refined to the algae Spirulina platensis, owing to its former use in the food industry and hence known safety in terms of a well-understood, non-toxic nature. A second material, cuprorivaite powder, also known as Egyptian blue pigment, was later simultaneously identified by two research groups as a second NIR-emitting material perfect for forensic application.[80,92] Cuprorivaite exhibits strong photoluminescence in the NIR region (λmax 910 nm) upon excitation at 630 nm, which corresponds to the B1g–Eg electronic transition.[92] The particle size of the powder was found to influence its colour intensity and so could be used to fine-tune the emissive properties. Use of a micronizing mill to effect a particle size of ~5 μm produced optimal emissive properties in the NIR region from cuprorivaite powder. NIR–NIR fluorescence has also been shown using cuprorivaite, wherein the B1g–B2g electronic transition is instead targeted (λmax 780 nm).[80] This material emits in the NIR region with an unusually high quantum yield of 10.5 %.[93]
Phosphorescence has similarly been explored as a basis for NIR powders because it may enable time-resolved imaging of surfaces to eliminate background emissions,[94,95] a strategy that has found similar use in biological systems.[36] This has been achieved using visibly emitting lanthanide phosphors, wherein a delay between excitation and imaging allows the more facile background emission to deplete, while the longer-lived phosphorescent fingermark emissions persist to the imaging step.[96,97] Zinc gallogermanate, Zn3Ga2Ge2O10 : 0.5 %Cr3+, has been trialled as a phosphorescent powder for this purpose.[98] This material can be excited using white light to provide long-lived NIR luminescent emission that spans a range of 650–1000 nm, while emitting an afterglow that lasts for up to 360 h.[99] Given this exceptionally long phosphorescent window, natural sunlight can readily be used to activate the material before photography, with visible contrast apparent two minutes after sunlight exposure.[98] This being said, the longer the delay before imaging, the greater the decay in the observable NIR emission.
Emergent Ln NIR Emitters for Fingermark Visualisation
The application of lanthanide-based NIR nanomaterials for fingerprinting is in its infancy, however, the same beneficial properties that recommend their use for bioimaging also apply to the forensic sciences. Upconversion nanorods comprised of NaYF4 doped with Yb, Er, and Gd have been applied to visualise fingerprints by triggering the green luminescent upconversion with a NIR laser (λ 980 nm). This technique was found to be effective across a wide range of surface types, including glass, ceramic, aluminium foil, stainless steel, and coins by simple physical adsorption of the powdered material.[100] Nanorods containing Nd3+ have similarly been used, with a Matryoshka-like core/shell/shell nanoparticle comprised of Tm, Yb, and Nd : Yb layers respectively.[101] Here, dual NIR emissions at 696 and 980 nm were observed upon excitation at 808 nm using a pump laser, providing both an upconversion and NIR–NIR luminescence approach to fingermark visualisation (Fig. 6). The attraction of the nanoparticles to the fingermark was attributed to electrostatic interactions, which were strengthened by chemical interactions between carboxyl groups of the powder and amine groups within the fingermark.
|
Upconversion nanoparticles containing europium or terbium have been used in conjunction with NIR laser sources to generate visible fluorescence based on harnessing two (or more) photons to emit higher-energy photons.[102] This approach was combined with a DNA aptamer that interacts with lysosomes, which is a cellular component found in fingermarks. The use of NIR wavelengths to produce visible luminescence minimises background interference owing to the low energy of excitation, necessitating the lanthanide-doped nanocrystals to provide the mechanism of upconversion. The UCNPs possessed excellent photostability in addition to the aforementioned beneficial photoproperties inherent to lanthanide phosphors. A second NIR-responsive upconversion nanomaterial that benefited from simple synthetic protocols was described by Mao et al.[54] This material (NaYF4 : Yb,Er) could be obtained as a highly pure, crystalline powder and was amenable to gram-scale synthesis. It produces an upconverted green fluorescence spectrum (λmax 542 nm) upon excitation at a wavelength of 980 nm, and it could be used as a powder to visualise the ridge patterns of latent fingermarks while negating background autofluorescence. It was also observed that crystal engineering principles could be applied to alter the crystal habit from the cubic α-phase to the hexagonal β-phase to enhance the fluorescent intensity, consistent with the strategy of targeting crystalline morphology to influence UCNP photoemission properties.[66]
These emergent lanthanide upconversion materials can be expected in future to outperform conventional powders, as well as other existing classes of nanoparticles, as a treatment for latent fingerprints.[54] One factor needing consideration before UCNP implementation in forensic science is the safety of this class of nanomaterial. Many of the materials discussed above contain toxic heavy metals, such as the cadmium used to sensitise Ln upconversion. Similarly, the processing of materials has been identified as an area of concern.[103] Safety issues have been flagged surrounding inhalation of the nanoparticles when used for fingermark dusting, including both quantum dots and lanthanide NIR powders.[81] These risks have been downplayed by other sources, with lanthanide NIR upconversion powders described as having low toxicity, or even no toxicity, based on the success of the biolabelling and cellular bioanalysis experiments described above given suitable surface modification.[55,104] The use of upconversion powders as liquid suspensions has been posited as a means of eliminating many of the hazards associated with airborne particulates, particularly if water can be used as the liquid medium. This strategy would likely necessitate functionalisation of the nanoparticle surfaces with hydrophobic groups to provide them with an affinity for the lipid fraction of fingermarks, building upon successful strategies employed for other fingermark reagents such as small particle reagents.[74]
Conclusion
This work reviews the recent developments in NIR-emissive Ln materials that have grown out of a need for improved biological and medical imaging, more efficient solar cells, and new materials for telecommunications. Concurrent to this, the benefits of visualising fingermarks using NIR emission and absorption have been recognised, and powerful forensic NIR light sources are more commonly found in the arsenal of the crime scene examiner. It is hoped that highlighting the complementarity of these two research endeavours will spur the application of new, cutting-edge Ln materials that will provide enhanced fingermark visualisation beyond present-day capabilities.
The lanthanide series, with their large Stokes shifts, long phosphorescent emission lifetimes, line-like emission profiles, high photostability, and low toxicity, are perfect candidates for forensic applications, as they have been for a plethora of other fields. Successfully implementing lanthanide UCNP materials capable of low-energy upconversion into the forensic sciences would have a transformative effect on current approaches to fingermark visualisation. Enhanced visualisation could be achieved in the form of less background interference, improved safety, and applicability to virtually any surface type.
Given the improving capabilities of both current and emerging UCNP materials, coupled with the recognition of NIR-emissive powders as the superior method for overcoming troublesome surfaces,[105] the arrival of the next generation of Ln materials tailored to forensic applications appears to be imminent.
Conflicts of Interest
The author declares no conflicts of interest.
Acknowledgements
The author is grateful to both the Royal Society (RG170176) and the Royal Society of Chemistry (RF18-4963) for financial support of this work.
References
[1] J.-C. G. Bünzli, S. V. Eliseeva, Chem. Sci. 2013, 4, 1939.| Crossref | GoogleScholarGoogle Scholar |
[2] S. Chen, A. Z. Weitemier, X. Zeng, L. M. He, X. Y. Wang, Y. Q. Tao, A. J. Y. Huang, Y. Hashimotodani, M. Kano, H. Iwasaki, L. K. Parajuli, S. Okabe, D. B. L. Teh, A. H. All, I. Tsutsui-Kimura, K. F. Tanaka, X. G. Liu, T. J. McHugh, Science 2018, 359, 679.
| Crossref | GoogleScholarGoogle Scholar | 29439241PubMed |
[3] G. E. Florence, W. J. Gee, Analyst 2018, 143, 3789.
| Crossref | GoogleScholarGoogle Scholar | 30014065PubMed |
[4] W. Zheng, P. Huang, D. Tu, E. Ma, H. Zhu, X. Chen, Chem. Soc. Rev. 2015, 44, 1379.
| Crossref | GoogleScholarGoogle Scholar | 25093303PubMed |
[5] A. Foucault-Collet, C. M. Shade, I. Nazarenko, S. Petoud, S. V. Eliseeva, Angew. Chem. Int. Ed. 2014, 53, 2927.
| Crossref | GoogleScholarGoogle Scholar |
[6] J.-C. G. Bünzli, C. Piguet, Chem. Soc. Rev. 2005, 34, 1048.
| Crossref | GoogleScholarGoogle Scholar |
[7] T. Jüstel, H. Nikol, C. Ronda, Angew. Chem. Int. Ed. 1998, 37, 3084.
| Crossref | GoogleScholarGoogle Scholar |
[8] M. J. F. Digonet, Rare Earth Doped Fiber Lasers and Amplifiers, 2nd edn 2001 (Marcel Dekker Inc.: New York, NY).
[9] J. Kido, Y. Okamoto, Chem. Rev. 2002, 102, 2357.
| Crossref | GoogleScholarGoogle Scholar | 12059271PubMed |
[10] S. SeethaLekshmi, A. R. Ramya, M. L. P. Reddy, S. Varughese, J. Photochem. Photobiol. Chem. 2017, 33, 109.
| Crossref | GoogleScholarGoogle Scholar |
[11] A. de Bettencourt-Dias, Dalton Trans. 2007, 2229.
| Crossref | GoogleScholarGoogle Scholar | 17534483PubMed |
[12] J. Heine, K. Müller-Buschbaum, Chem. Soc. Rev. 2013, 42, 9232.
| Crossref | GoogleScholarGoogle Scholar | 24077361PubMed |
[13] K. Binnemans, Chem. Rev. 2009, 109, 4283.
| Crossref | GoogleScholarGoogle Scholar | 19650663PubMed |
[14] L. A. Galán, B. L. Reid, S. Stagni, A. N. Sobolev, B. W. Skelton, E. G. Moore, G. S. Hanan, E. Zysman-Colman, M. I. Ogden, M. Massi, Dalton Trans. 2018, 47, 7956.
| Crossref | GoogleScholarGoogle Scholar |
[15] S. Sato, M. Wada, Bull. Chem. Soc. Jpn. 1970, 43, 1955.
| Crossref | GoogleScholarGoogle Scholar |
[16] J. Li, H. Li, P. Yan, P. Chen, G. Hou, G. Li, Inorg. Chem. 2012, 51, 5050.
| Crossref | GoogleScholarGoogle Scholar | 22519622PubMed |
[17] D. B. A. Raj, S. Biju, M. L. P. Reddy, J. Mater. Chem. 2009, 19, 7976.
| Crossref | GoogleScholarGoogle Scholar |
[18] P. C. Andrews, W. J. Gee, P. C. Junk, M. Massi, New J. Chem. 2013, 37, 35.
| Crossref | GoogleScholarGoogle Scholar |
[19] T. M. George, S. Varughese, M. L. P. Reddy, RSC Adv. 2016, 6, 69509.
| Crossref | GoogleScholarGoogle Scholar |
[20] M. L. P. Reddy, V. Divya, R. Pavithran, Dalton Trans. 2013, 42, 15249.
| Crossref | GoogleScholarGoogle Scholar |
[21] G. Zucchi, O. Maury, P. Thuéry, M. Ephritikhine, Inorg. Chem. 2008, 47, 10398.
| Crossref | GoogleScholarGoogle Scholar | 18847254PubMed |
[22] B. L. Reid, S. Stagni, J. M. Malicka, M. Cocchi, G. S. Hanan, M. I. Ogden, M. Massi, Chem. Commun. 2014, 50, 11580.
| Crossref | GoogleScholarGoogle Scholar |
[23] B. L. Reid, S. Stagni, J. M. Malicka, M. Cocchi, A. N. Sobolev, B. W. Skelton, E. G. Moore, G. S. Hanan, M. I. Ogden, M. Massi, Chem. – Eur. J. 2015, 21, 18354.
| Crossref | GoogleScholarGoogle Scholar | 26511032PubMed |
[24] S. Biju, Y. K. Eom, J.-C. Bünzli, H. K. Kim, J. Mater. Chem. C 2013, 1, 6935.
| Crossref | GoogleScholarGoogle Scholar |
[25] T. Zhang, X. Zhu, W.-K. Wong, H.-L. Tam, W.-Y. Wong, Chem. – Eur. J. 2013, 19, 739.
| Crossref | GoogleScholarGoogle Scholar | 23165692PubMed |
[26] T. N. Nguyen, G. Capano, A. Gładysiak, F. M. Ebrahim, S. V. Eliseeva, A. Chidambaram, B. Valizadeh, S. Petoud, B. Smit, K. C. Stylianoa, Chem. Commun. 2018, 54, 6816.
| Crossref | GoogleScholarGoogle Scholar |
[27] L.-H. Liu, X.-T. Qiu, Y.-J. Wang, Q. Shi, Y.-Q. Sun, Y.-P. Chen, Dalton Trans. 2017, 46, 12106.
| Crossref | GoogleScholarGoogle Scholar | 28858344PubMed |
[28] X. Lian, D. Zhao, Y. Cui, Y. Yang, G. Qian, Chem. Commun. 2015, 51, 17676.
| Crossref | GoogleScholarGoogle Scholar |
[29] A. T. Bui, A. Grichine, S. Brasselet, A. Duperray, C. Andraud, O. Maury, Chem. – Eur. J. 2015, 21, 17757.
| Crossref | GoogleScholarGoogle Scholar | 26489885PubMed |
[30] J. V. Frangioni, Curr. Opin. Chem. Biol. 2003, 7, 626.
| Crossref | GoogleScholarGoogle Scholar | 14580568PubMed |
[31] D. K. Sinha, P. Neveu, N. Gagey, I. Aujard, C. Benbrahim-Bouzidi, T. Le Saux, C. Rampon, C. Gauron, B. Goetz, S. Dubruille, M. Baaden, M. Volovitch, D. Bensimon, S. Vriz, L. Jullien, ChemBioChem 2010, 11, 653.
| Crossref | GoogleScholarGoogle Scholar | 20187057PubMed |
[32] S. J. Butler, M. Delbianco, L. Lamarque, B. K. McMahon, E. R. Neil, R. Pal, S. Parker, J. W. Walton, J. M. Zwier, Dalton Trans. 2015, 44, 4791.
| Crossref | GoogleScholarGoogle Scholar | 25341077PubMed |
[33] A. D’Aléo, A. Bourdolle, S. Brustlein, T. Fauquier, A. Grichine, A. Duperray, P. L. Baldeck, C. Andraud, S. Brasselet, O. Maudy, Angew. Chem. Int. Ed. 2012, 51, 6622.
| Crossref | GoogleScholarGoogle Scholar |
[34] A. Foucault-Collet, K. A. Gogick, K. A. White, S. Villette, A. Pallier, G. Collet, C. Kieda, T. Li, S. J. Geib, N. L. Rosi, S. Petoud, Proc. Natl. Acad. Sci. USA 2013, 110, 17199.
| Crossref | GoogleScholarGoogle Scholar | 24108356PubMed |
[35] R. Weissleder, V. Ntziachristos, Nat. Med. 2003, 9, 123.
| Crossref | GoogleScholarGoogle Scholar | 12514725PubMed |
[36] W. Zheng, D. Tu, P. Huang, S. Zhou, Z. Chen, X. Chen, Chem. Commun. 2015, 51, 4129.
| Crossref | GoogleScholarGoogle Scholar |
[37] W. P. Fan, W. B. Bu, J. L. Shi, Adv. Mater. 2016, 28, 3987.
| Crossref | GoogleScholarGoogle Scholar |
[38] S. Wu, H. J. Butt, Adv. Mater. 2016, 28, 1208.
| Crossref | GoogleScholarGoogle Scholar | 26389516PubMed |
[39] Y. I. Park, K. T. Lee, Y. D. Suh, T. Hyeon, Chem. Soc. Rev. 2015, 44, 1302.
| Crossref | GoogleScholarGoogle Scholar | 25042637PubMed |
[40] D. M. Yang, P. A. Ma, Z. Y. Hou, Z. Y. Cheng, C. X. Li, J. Lin, Chem. Soc. Rev. 2015, 44, 1416.
| Crossref | GoogleScholarGoogle Scholar |
[41] M. Wang, G. Abbineni, A. Clevenger, C. Mao, S. Xu, Nanomedicine: Nanotech. Biol. Med. 2011, 7, 710.
| Crossref | GoogleScholarGoogle Scholar |
[42] J. Zhou, Q. Liu, W. Feng, Y. Sun, F. Li, Chem. Rev. 2015, 115, 395.
| Crossref | GoogleScholarGoogle Scholar | 25492128PubMed |
[43] J. Liu, H. Deng, Z. Huang, Y. Zhang, D. Chen, Y. Shao, Phys. Chem. Chem. Phys. 2015, 17, 15412.
| Crossref | GoogleScholarGoogle Scholar | 26006337PubMed |
[44] L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J.-C. G. Bünzli, A. Hauser, C. Piguet, Angew. Chem. Int. Ed. 2011, 50, 4108.
| Crossref | GoogleScholarGoogle Scholar |
[45] X. Chen, D. Peng, Q. Ju, F. Wang, Chem. Soc. Rev. 2015, 44, 1318.
| Crossref | GoogleScholarGoogle Scholar | 25058157PubMed |
[46] B. Zhou, B. Shi, D. Jin, X. Liu, Nat. Nanotechnol. 2015, 10, 924.
| Crossref | GoogleScholarGoogle Scholar | 26530022PubMed |
[47] G. Chen, H. Qiu, P. N. Prasad, X. Chen, Chem. Rev. 2014, 114, 5161.
| Crossref | GoogleScholarGoogle Scholar | 24605868PubMed |
[48] C. Chen, C. Li, Z. Shi, Adv. Sci. 2016, 3, 1600029.
| Crossref | GoogleScholarGoogle Scholar |
[49] J. F. Suyver, A. Aebischer, D. Biner, P. Gerner, J. Grimm, S. Heer, K. W. Krämer, C. Reinhard, H. U. Güdel, Opt. Mater. 2005, 27, 1111.
| Crossref | GoogleScholarGoogle Scholar |
[50] J. Shen, G. Y. Chen, A. M. Vu, W. Fan, O. S. Bilsel, C. C. Chang, G. Han, Adv. Opt. Mater. 2013, 1, 644.
| Crossref | GoogleScholarGoogle Scholar |
[51] Y. T. Zhong, G. Tian, Z. J. Gu, Y. J. Yang, L. Gu, Y. L. Zhao, Y. Ma, J. N. Yao, Adv. Mater. 2014, 26, 2831.
| Crossref | GoogleScholarGoogle Scholar |
[52] F. Auzel, Chem. Rev. 2004, 104, 139.
| Crossref | GoogleScholarGoogle Scholar | 14719973PubMed |
[53] G. Liu, B. Jacquier, Spectroscopic Properties of Rare Earths in Optical Materials 2005 (Springer: Berlin).
[54] M. Wang, Y. Zhu, C. Mao, Langmuir 2015, 31, 7084.
| Crossref | GoogleScholarGoogle Scholar | 26089129PubMed |
[55] M. Wang, M. Li, A. Yu, Y. Zhu, M. Yang, C. Mao, Adv. Funct. Mater. 2017, 27, 1606243.
| Crossref | GoogleScholarGoogle Scholar | 29657570PubMed |
[56] K. W. Krämer, D. Biner, G. Frei, H. U. Gudel, M. P. Hehlen, S. R. Luthi, Chem. Mater. 2004, 16, 1244.
| Crossref | GoogleScholarGoogle Scholar |
[57] R. Ma, R. Shimmon, A. McDonagh, P. Maynard, C. Lennard, C. Roux, Forensic Sci. Int. 2012, 217, e23.
| Crossref | GoogleScholarGoogle Scholar | 22047749PubMed |
[58] G. Y. Chen, H. C. Liu, G. Somesfalean, Y. Q. Sheng, H. J. Liang, Z. G. Zhang, Q. Sun, F. P. Wang, Appl. Phys. Lett. 2008, 92, 113114.
| Crossref | GoogleScholarGoogle Scholar |
[59] Y. F. Bai, Y. X. Wang, K. Yang, X. R. Zhang, G. Y. Peng, Y. L. Song, Z. Y. Pan, C. H. J. Wang, PhysChemComm 2008, 112, 12259.
[60] G. Y. Chen, H. C. Liu, H. J. Liang, G. Somesfalean, Z. G. Zhang, J. Phys. Chem. C 2008, 112, 12030.
| Crossref | GoogleScholarGoogle Scholar |
[61] Y. F. Bai, Y. X. Wang, K. Yang, X. R. Zhang, Y. L. Song, C. H. Wang, Opt. Commun. 2008, 281, 5448.
| Crossref | GoogleScholarGoogle Scholar |
[62] Z. L. Li, B. S. Wang, L. C. Xing, S. L. Liu, N. Tan, S. G. Xiao, J. W. Ding, Chin. Opt. Lett. 2012, 10, 081602.
| Crossref | GoogleScholarGoogle Scholar |
[63] W. Q. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov, J. C. Hummelen, Nat. Photonics 2012, 6, 560.
| Crossref | GoogleScholarGoogle Scholar |
[64] F. Wang, R. R. Deng, J. Wang, Q. X. Wang, Y. Han, H. M. Zhu, X. Y. Chen, X. G. Liu, Nat. Mater. 2011, 10, 968.
| Crossref | GoogleScholarGoogle Scholar | 22019945PubMed |
[65] J. W. Wang, P. A. Tanner, J. Am. Chem. Soc. 2010, 132, 947.
| Crossref | GoogleScholarGoogle Scholar |
[66] F. Wang, X. G. Liu, Acc. Chem. Res. 2014, 47, 1378.
| Crossref | GoogleScholarGoogle Scholar | 24611606PubMed |
[67] X. M. Li, F. Zhang, D. Y. Zhao, Chem. Soc. Rev. 2015, 44, 1346.
| Crossref | GoogleScholarGoogle Scholar |
[68] G. Chen, H. Ågren, T. Y. Ohulchanskyy, P. N. Prasad, Chem. Soc. Rev. 2015, 44, 1680.
| Crossref | GoogleScholarGoogle Scholar | 25335878PubMed |
[69] Y. Suffren, D. Zare, S. V. Eliseeva, L. Guénée, H. Nozary, T. Lathion, L. Aboshyan-Sorgho, S. Petoud, A. Hauser, C. Piguet, J. Phys. Chem. C 2013, 117, 26957.
| Crossref | GoogleScholarGoogle Scholar |
[70] D. Zare, Y. Suffren, L. Guénée, S. V. Eliseeva, H. Nozary, L. Aboshyan-Sorgho, S. Petoud, A. Hauser, C. Piguet, Dalton Trans. 2015, 44, 2529.
| Crossref | GoogleScholarGoogle Scholar | 25357092PubMed |
[71] D. Zare, Y. Suffren, H. Nozary, A. Hauser, C. Piguet, Angew. Chem. Int. Ed. 2017, 56, 14612.
| Crossref | GoogleScholarGoogle Scholar |
[72] H. Suzuki, Y. Nishida, S. Hoshino, Mol. Cryst. Liq. Cryst. 2003, 406, 27.
| Crossref | GoogleScholarGoogle Scholar |
[73] B. Su, Anal. Bioanal. Chem. 2016, 408, 2781.
| Crossref | GoogleScholarGoogle Scholar | 26781104PubMed |
[74] C. Champod, C. J. Lennard, P. Margot, M. Stoilovic, Fingerprints and Other Ridge Skin Impressions, 2nd edn 2016 (CRC Press: Boca Raton, FL).
[75] G. S. Sodhi, J. Kaur, Forensic Sci. Int. 2001, 120, 172.
| Crossref | GoogleScholarGoogle Scholar | 11473799PubMed |
[76] R. S. P. King, P. M. Hallett, D. Foster, Forensic Sci. Int. 2015, 249, e21.
| Crossref | GoogleScholarGoogle Scholar |
[77] L. W. Hayball, Advanced Infrared Photography Handbook 2001 (Amherst Media: Buffalo, NY).
[78] P. Maynard, J. Jenkins, C. Edey, G. Payne, C. Lennard, A. McDonagh, C. Roux, Aus. J. Forensic Sci. 2009, 41, 43.
| Crossref | GoogleScholarGoogle Scholar |
[79] S. Chadwick, P. Maynard, P. Kirkbride, C. Lennard, X. Spindler, C. Roux, J. Forensic Sci. 2011, 56, 1505.
| Crossref | GoogleScholarGoogle Scholar | 21790599PubMed |
[80] R. S. P. King, P. M. Hallett, D. Foster, Forensic Sci. Int. 2016, 262, e28.
| Crossref | GoogleScholarGoogle Scholar |
[81] A. Bécue, Anal. Methods 2016, 8, 7983.
| Crossref | GoogleScholarGoogle Scholar |
[82] M. Algarra, J. Jiménez-Jiménez, M. S. Miranda, B. B. Campos, R. Moreno-Tost, E. Rodriguez-Castellón, J. C. G. Esteves da Silva, Surf. Interface Anal. 2013, 45, 612.
| Crossref | GoogleScholarGoogle Scholar |
[83] J. Dilag, K. Kobus, A. V. Ellis, Forensic Sci. Int. 2009, 187, 97.
| Crossref | GoogleScholarGoogle Scholar | 19356872PubMed |
[84] J. Dilag, K. Kobus, A. V. Ellis, Forensic Sci. Int. 2013, 228, 105.
| Crossref | GoogleScholarGoogle Scholar | 23597747PubMed |
[85] T. Yang, X. Guo, H. Wang, S. Fu, Y. Wen, H. Yang, Biosens. Bioelectron. 2015, 68, 350.
| Crossref | GoogleScholarGoogle Scholar | 25603400PubMed |
[86] S. K. Singh, K. Kumar, S. B. Rai, Appl. Phys. B 2009, 94, 165.
| Crossref | GoogleScholarGoogle Scholar |
[87] R. Ma, E. Bullock, P. Maynard, B. Reedy, R. Shimmon, C. Lennard, C. Roux, A. McDonagh, Forensic Sci. Int. 2011, 207, 145.
| Crossref | GoogleScholarGoogle Scholar | 20980110PubMed |
[88] H.-H. Xie, Q. Wen, H. Huang, T.-Y. Sun, P. Li, Y. Li, X.-F. Yu, Q.-Q. Wang, RSC Adv. 2015, 5, 79525.
| Crossref | GoogleScholarGoogle Scholar |
[89] M. Wang, M. Li, M. Yang, X. Zhang, A. Yu, Y. Zhu, P. Qiu, C. Mao, Nano Res. 2015, 8, 1800.
| Crossref | GoogleScholarGoogle Scholar | 27818741PubMed |
[90] S. P. Tiwari, K. Kumar, V. K. Rai, J. Appl. Phys. 2015, 118, 183109.
| Crossref | GoogleScholarGoogle Scholar |
[91] S. P. Tiwari, K. Kumar, V. K. Rai, Appl. Phys. B 2015, 121, 221.
| Crossref | GoogleScholarGoogle Scholar |
[92] B. Errington, G. Lawson, S. W. Lewis, G. D. Smith, Dyes Pigments 2016, 132, 310.
| Crossref | GoogleScholarGoogle Scholar |
[93] G. Accorsi, G. Verri, M. Bolognesi, N. Armaroli, C. Clementi, C. Miliani, A. Romani, Chem. Commun. 2009, 3392.
| Crossref | GoogleScholarGoogle Scholar |
[94] E. R. Menzel, J. Forensic Sci. 1979, 24, 582.
[95] E. R. Menzel, Anal. Chem. 1989, 61, 557A.
| Crossref | GoogleScholarGoogle Scholar |
[96] E. R. Menzel, Fingerpr. Whorld 1997, 45.
[97] X. Xiong, X. Yuan, J. Song, G. Yin, Appl. Spectrosc. 2016, 70, 995.
| Crossref | GoogleScholarGoogle Scholar | 27076516PubMed |
[98] R. S. P. King, D. A. Skros, Forensic Sci. Int. 2017, 276, e35.
| Crossref | GoogleScholarGoogle Scholar |
[99] Z. Pan, Y.-Y. Lu, F. Liu, Nat. Mater. 2012, 11, 58.
| Crossref | GoogleScholarGoogle Scholar |
[100] B.-Y. Li, X.-L. Zhang, L.-Y. Zhang, T.-T. Wang, L. Li, C.-G. Wang, Z.-M. Su, Dyes Pigments 2016, 134, 178.
| Crossref | GoogleScholarGoogle Scholar |
[101] J. Li, X. Zhu, M. Xue, W. Feng, R. Ma, F. Li, Inorg. Chem. 2016, 55, 10278.
| Crossref | GoogleScholarGoogle Scholar | 27684997PubMed |
[102] J. Wang, T. Wei, X. Li, B. Zhang, J. Wang, C. Huang, Q. Yuan, Angew. Chem. Int. Ed. 2014, 53, 1616.
| Crossref | GoogleScholarGoogle Scholar |
[103] S. Singh, H. S. Nalwa, J. Nanosci. Nanotechnol. 2007, 7, 3048.
| Crossref | GoogleScholarGoogle Scholar | 18019130PubMed |
[104] Y. Sun, W. Feng, P. Y. Yang, C. H. Huang, F. Y. Li, Chem. Soc. Rev. 2015, 44, 1509.
| Crossref | GoogleScholarGoogle Scholar | 25113504PubMed |
[105] R. P. Downham, E. R. Brewer, R. S. P. King, A. M. Luscombe, V. G. Sears, Forensic Sci. Int. 2017, 275, 30.
| Crossref | GoogleScholarGoogle Scholar | 28292656PubMed |