

Ruthenium Alkynyl Complexes in Non-Linear Optics*
Mark G. HumphreyResearch School of Chemistry, Australian National University, Canberra, ACT 2601, Australia. Email: mark.humphrey@anu.edu.au
Australian Journal of Chemistry 71(10) 731-742 https://doi.org/10.1071/CH18325
Submitted: 6 July 2018 Accepted: 3 August 2018 Published: 3 September 2018
Abstract
Non-linear optical (NLO) materials are able to modify the propagation characteristics of light. Such materials have a range of potential applications in advanced technologies and are therefore of considerable interest. This account summarizes the development of one class of organometallics as potential NLO materials, namely ruthenium alkynyl complexes. These are available in high yields by straightforward synthetic procedures and have good thermal and environmental stability. In studies ranging from small molecules (molecular weights ~1000) to second-generation dendrimers (with molecular weights of more than 20000), the author’s group and collaborators have assayed the NLO effects in complexes with a variety of ‘multipolar’ charge distributions (dipolar, quadrupolar, octupolar), revealing that ruthenium alkynyl complexes can be engineered to display record and near-record values of the parameters responsible for various interesting NLO effects. In particular, recent studies driven by the current focus on optimizing molecular multiphoton absorption cross-sections have afforded several examples with world-record values of these key coefficients. The author’s group has also shown that the fully reversible redox processes undergone by many ruthenium alkynyl complexes are a distinctive feature that can be exploited to afford molecular NLO switches, because the different and reversibly accessible redox forms of the complexes exhibit measurably different NLO responses. This unique type of switching has been extended in two ways to afford molecular switches with multiple accessible NLO states. First, ruthenium alkynyl complexes have been subjected to various ‘orthogonal’ (independent) switching stimuli (specifically oxidation–reduction, protonation–deprotonation, and photoisomerization), affording complexes that function as NLO switches with up to six distinct NLO states. Second, heterobimetallic complexes coupling ruthenium alkynyl and iron alkynyl centres have been prepared that exhibit multiple redox-accessible NLO states.
Introduction
The field of non-linear optics was launched following the development of the laser, which afforded ready access to the high-intensity light needed to achieve observable non-linear effects. Materials that possess the requisite non-linear optical (NLO) properties can modify the propagation characteristics of light (frequency, amplitude, path, phase, polarization, etc.) and, as a result, they are of significant interest for applications in laser technology, data storage, telecommunications, optical signal and image processing, biological imaging, and nanofabrication. Unlike electronics, where silicon is the material of choice for most applications (excluding throwaway devices), there is thus far no broadly useful material for photonics applications. A wide variety of materials have been assayed for NLO applications, including inorganic salts, organic molecules, polymers, liquid crystals, and composites, but molecular organic, inorganic, and organometallic compounds have attracted special interest because of the ease with which their composition can be modified, and consequently structure–property relationships established from which to optimize behaviour, as well as the large values of the key molecular NLO parameters that have been obtained for some examples. This account summarizes some of our studies exploring the NLO properties of organometallics, with a particular focus on ruthenium alkynyl complexes.
The theoretical background to the NLO materials field has been presented elsewhere,[1] and will not be reproduced in detail here; rather, a short introduction will be presented to clarify the subsequent discussion. Briefly, an intense electric field Eloc incident on a molecule can distort its electron density distribution, and the resultant change in the dipole moment μ0 is given by:
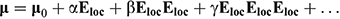
where the tensors α, β, and γ are the linear polarizability (a 3 × 3 matrix), the quadratic hyperpolarizability (a 3 × 3 × 3 matrix), and the cubic hyperpolarizability (a 3 × 3 × 3 × 3 matrix) respectively. This may seem complicated, but many tensor components are equivalent by symmetry rules (or equal to zero), resulting in considerable simplification, while polarizabilities are invariant with respect to all point group symmetry operations, so all the components of β vanish in centrosymmetric point groups. A similar relation describes polarization at the macroscopic level:

where P is the macroscopic polarization, P0 is the intrinsic polarization, χ(n) are the n-th order susceptibilities, and E is the electric field, but the focus of the present account is on NLO effects at the molecular level.
Because the electric field of a light wave oscillates and can be written as E(t) = E0cos(ωt) where E(t) is the time-dependent electric field, it is immediately apparent from considering the trigonometric expansions cos2(ωt) = 1/2 + 1/2cos(2ωt) and cos3(ωt) = 3/4cos(ωt) + 1/4cos(3ωt) that the non-linear terms in the equation above introduce contributions at double and triple the fundamental frequency (i.e. second-harmonic generation (SHG) and third-harmonic generation (THG)), as well as a time-independent (direct current (DC)) contribution. A variety of other interesting effects are also possible; the key requirement is to generate materials that have sufficiently large NLO coefficients, while being stable to processing and device operating conditions.
A range of techniques has been employed to measure NLO properties of materials; detailed descriptions can be found elsewhere.[1d] Many of the early reports of NLO effects employed the Kurtz powder technique, in which the second-harmonic light scattered from a microcrystalline powder is assessed. This has the advantage of simplicity, and it has indeed been deployed to assess the bulk material second-order NLO activity of vinylidene complexes[2] and alkynyl complexes[3] relevant to the present account, but it is generally just a qualitative indication of NLO activity, and it has been largely supplanted by more quantitative techniques measuring NLO performance at the molecular level. The quadratic NLO properties of metal alkynyl complexes have mostly been assessed using the hyper-Rayleigh scattering technique (HRS).[4] HRS involves focussing an incident beam on a solution of the sample. A concave mirror and collecting lens then collect the scattered light. The light is filtered to remove the fundamental radiation, leaving the second-harmonic light (Fig. 1). HRS replaced electric field-induced SHG in the 1990s as the method of choice because (i) it is simpler (a DC field is not needed, and there is no need to measure the dipole moment or the cubic non-linearity); (ii) it can sample the off-diagonal tensor components of the quadratic non-linearity; and (iii) it is applicable to a broader range of compounds (ionic compounds, octupolar compounds). It has a couple of shortcomings: (i) it does not afford the sign of β, but this is not a major problem because it is usually obvious, and (ii) any fluorescence contributions need to be removed.

|
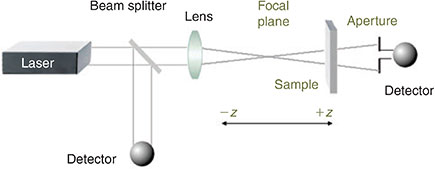
Cubic NLO properties of metal alkynyl complexes have mostly been assessed using the Z-scan technique,[5] which largely displaced degenerate four-wave mixing and other procedures in the 1990s owing to its experimental simplicity (a single-beam configuration giving both the real and imaginary components of χ(3) and consequently γ). Z-scan involves moving a solution of the sample on a motorized stage through the focal plane of a laser. The self-focussing behaviour and intensity-dependent absorption are thereby assessed (Fig. 2). Z-scan has the disadvantage of not affording temporal information about the NLO responses, but this has not hindered its broad implementation.
|
Although it was not the first report of the NLO properties of organometallic complexes,[6] the seminal study by Green et al. of SHG effects exhibited by powders of a certain ferrocenyl complex alerted the organometallic chemistry community to the potential of such complexes in this then-nascent field.[7] A wide variety of organometallics (as well as organics and coordination complexes: see above) were assessed in the 1990s, affording some design rules for efficient NLO-active compounds.[8] The need for both sufficient activity and sufficient stability for putative applications gradually focussed the organometallic field on metallocenyl (particularly ferrocenyl) and metal alkynyl complexes. An early report identified a potential shortcoming with metallocenyl complexes: the intra-chromophore charge transfer contributing to the NLO responses is orthogonal to the metal-to-cyclopentadienyl charge-transfer axis (Fig. 3, left), a potentially unfavourable arrangement,[9] although subsequent more-detailed studies have suggested that the key transitions in these metallocenyl complexes consist of a low-energy metal-to-acceptor transition and a higher-energy chromophore π-system-to-acceptor transition.[10] In contrast, metal alkynyl complexes feature the ligated metal collinear with the intra-alkynyl ligand charge transfer (Fig. 3, right), potentially affording the possibility of stronger coupling of the metal, and thereby enhanced optical non-linearities.[11]

|
Metal alkynyl complexes have commanded significant attention from organometallic chemists since the initial report of nickel and copper alkynyls by Nast in 1953.[12] Early studies of metal alkynyl complexes focussed on synthesis, structure, and bonding,[13] and metal alkynyl complexes now exist for essentially all of the transition metals. Subsequently, the reactivity of these complexes came to the fore, with reports affording a plethora of derivative ligands (vinylidenes, carbenes, etc.),[14] and with ensuing work extending these studies to applications in catalysis.[15] More recently, materials applications of metal alkynyl complexes have been a focus, owing to their electron-rich and π-delocalizable nature.[16] In particular, in addition to the aforementioned interest in non-linear optics, their potential use in molecular electronics has been actively pursued.[17]
Ruthenium arguably sits in a Goldilocks zone in the Periodic Table. Ruthenium’s finely tuned balance of properties in catalysis applications led to Wilkinson describing it as ‘an element for the connoisseur’.[18] Ruthenium also possesses an optimal combination of properties for NLO applications.[19] Early studies revealed that quadratic optical non-linearities for metal alkynyl complexes increase on proceeding from 14-electron gold alkynyl complexes[20] to 18-electron complexes, and increase further on replacing a less readily oxidized, less polarizable 3d metal such as nickel[20b,21] with a more readily oxidized, more readily polarizable 4d metal such as ruthenium (Fig. 4).[20b,22] Consistent with these trends, the non-linearities also increase on proceeding from 3d iron to 4d ruthenium and then 5d osmium in analogous complexes,[23] but the relative kinetic inertness of the last-mentioned metal has stymied development of its chemistry and applications. Ruthenium is therefore the metal of choice in non-linear optics, as it is in many other applications. This account summarizes progress in the area of ruthenium alkynyl complexes in non-linear optics, with a particular focus on research in the author’s laboratory, and with an emphasis on molecular rather than bulk material studies. Computational studies of the NLO properties of ruthenium alkynyl complexes employing semi-empirical and density functional theory approaches have also been undertaken,[22a–c,24] but it remains the case that benchmarking the computational data against experimental data is necessary, and so the focus of this account is experimentally derived NLO parameters.

|
Quadratic NLO Properties of Ruthenium Alkynyl Complexes
Simple molecular design rules for efficient NLO-active materials were developed with small organic molecules and then propagated to the coordination, organometallic, and macromolecular realms. Studies correlating molecular structure with quadratic NLO properties of organic compounds have been summarized extensively elsewhere,[25] so only relevant key outcomes will be mentioned here. One useful relationship derived from perturbation theory for molecules with a single charge-transfer excited state that dominates the quadratic hyperpolarizability is:

This describes β in terms of the difference in excited-state and ground-state dipole moment μee – μgg, transition dipole moment μge, and optical absorption energy Ege of the key low-energy charge-transfer transition, so compounds exhibiting intense (large μge) charge-transfer transitions (large μee – μgg) at long wavelength (small Ege) are suggested to possess large values of β. Not surprisingly, the most efficient purely organic quadratic NLO materials are dipolar with a donor–bridge–acceptor composition, and this is also true of organometallics. The most NLO-efficient dipolar metal alkynyl complexes have generally featured an electron-rich ligated metal donor linked by an arylalkynyl bridge to an organic acceptor group, although the metal centre has also been employed in the bridge[26] and as an acceptor group.[27] With the ligated metal unit as donor in a donor–bridge–acceptor composition, trends in the key coefficient βHRS on varying the arylalkynyl ligand π-bridge length, solubilizing groups, aryl linking unit stereochemistry and composition, and acceptor group were established and rationalized in the 1990s and 2000s, affording design rules for efficient metal alkynyl-based NLO molecular materials (see Fig. 5 for examples of the structure–NLO property outcomes with certain gold complexes: β increases on incorporation of acceptor group, increasing the strength of the acceptor, progression from phenylene to heterocyclic unit, lengthening the π-system, progression from Z- to E-ene stereochemistry, and progression from yne- to imino-, ene-, and azo-linkage in the bridge unit).[20a,28]

|
Such changes involve the organic arylalkynyl ligand, but the greater interest with organometallics lies in the effect of structural modifications at the ligated metal centre. It is well established that the chemical and physical properties of transition metal complexes can usually be tuned by modifying the co-ligands comprising the coordination sphere. With organometallic complexes, this is a procedure with an enormous number of applications in catalysis,[29] but it can also be employed as an attractive method to modify NLO properties, and this has been demonstrated with ruthenium alkynyl complexes. For example, with complexes in which the ligated metal functions as a donor group in a donor–π-bridge–acceptor construction, examples containing the comparatively electron-withdrawing co-ligand carbonyl, not surprisingly, have significantly lower non-linearities than those possessing the electron-donating co-ligands PPh3, 1,2-bis(diphenylphosphino)ethane (dppe), or 1,1′-bis(diphenylphosphino)ferrocene (dppf) (Table 1).[22b,22c,23,24c] π-Electron delocalization facilitated by the co-ligand is another important consideration, and in some cases, this can prove more important than donor strength; progression from complexes incorporating the co-ligand PMe3 (a strong donor) to analogues with PPh3 (with greater π-delocalization possibilities) resulted in an increase in quadratic NLO response (Table 1).[22b,22c]

|
NLO effects can be understood in terms of the polarizing electric fields modifying the ground state of a molecule and thereby generating a new state corresponding to the instantaneous mixing of excited states into the ground state.[30] When the excited states involve substantial charge displacement from the ground state, such as with states resulting from charge transfer, the NLO effects can be significant. Metal arylalkynyl complexes exhibit strong charge transfer along the metal–alkynyl axis when excited, and such complexes are therefore of interest. However, although complexes possessing arylalkynyl ligands have been designed to exhibit significant optical non-linearities, they lose aromatic stabilization energy when excited to the key charge-transfer state (Fig. 6, left). To counter this deleterious outcome, complexes containing indoanilinoalkynyl ligands have been synthesized, because they possess a charge-transfer excited state that can be accessed without loss of aromatic stabilization energy (Fig. 6, right); pleasingly, these exhibit significant non-linearities.[31]

|
The non-linearities of metal alkynyl complexes with a donor–π-bridge–acceptor composition can be tuned by modification of any of the three components. The ligated metal has functioned as a donor in most of examples, with the non-linearity being modified by metal replacement, co-ligand modification (see above), and acceptor group replacement (progressing from no acceptor (H) to a weak acceptor (CHO) and then to a strong acceptor (NO2) at a 4-substituted arylalkynyl ligand results in increased quadratic optical non-linearity: Fig. 5).[20a,21,22c,28,32] The NLO merit has also been tuned by bridge modification. In addition to the various bridge modifications summarized above (Fig. 5), bridge lengthening has attracted attention, and it has been shown that quadratic non-linearities do not increase indefinitely, but instead they tend to saturate at a certain length (Table 2).[24e,33] Bridge ‘broadening’ has also been explored, with non-linearities increasing on proceeding from a 1,4-phenylenyl to 1,4-naphthalenyl bridge.[24h]

|
The aforementioned dipolar metal alkynyl complexes can be exceptionally NLO-efficient: quadratic non-linearities (resonance-enhanced and two-level corrected) for some of these alkynyl complexes are among the largest for organometallic compounds thus far.[33d,34] However, dipolar molecules suffer from an NLO efficiency–optical transparency trade-off (i.e. increasing non-linearity is associated with a red shift of the important linear optical absorption band corresponding to strong charge transfer, and this reduces the spectral range available to exploit the NLO properties). This problem has been met and addressed with organic molecules. Zyss and coworkers showed that because higher-order multipoles may also contribute to optical non-linearities, octupolar organic compounds can possess significant NLO properties while maintaining optimum transparency.[35] Similarly, metal alkynyl complexes derived from 1,3,5-triethynylbenzene have a 3-fold symmetry-axis and are octupolar, and ruthenium-containing examples have been studied.[36] Examples thus far have lacked strongly polarizing groups at the central arene ring and quadratic non-linearities are consequently small, but are non-zero, demonstrating the potential of this approach. In some instances, complexes corresponding to an ‘arm’ of the octupolar star complexes have also been examined, and the data contrasted with those from the octupolar examples to demonstrate a ‘dimensional evolution’ of NLO properties (progressing from the one-dimensional linear ‘arm’ complex to the two-dimensional octupolar complex).[36c,37]
Cubic NLO Properties of Ruthenium Alkynyl Complexes
Cubic NLO properties of ruthenium alkynyl complexes were first reported in the mid-to-late 1990s,[36b,38] and this area continues to attract interest.[11e,39] Similarly to the situation with quadratic NLO properties, structure-cubic NLO property correlations have been established with organic molecules and propagated to the organometallic realm. In general, and in contrast to quadratic NLO properties, the structure–property relationships are not clear-cut, with the exception (up to a certain size) of a general dependence on the size of the π-system. As is the case with quadratic NLO properties, there is a useful relationship to assist cubic NLO materials design that has been derived from perturbation theory:

where μee′ and μge are transition dipole moments, μgg is the ground-state dipole moment, μee is the excited-state dipole moment, and Ege and Ege′ are optical absorption energies. The third component of this expression has μge and (μee – μgg) in the numerator and Ege in the denominator, similarly to the expression for β, and indeed, in some cases, trends in the global cubic NLO parameter γ have been found to be similar to those seen with βHRS (e.g. varying the nature of the arylalkynyl ligand). The dependence on the size of the π-system focussed attention on complexes with extended π-delocalizable alkynyl ligands such as ruthenium-functionalized oligo(phenyleneethynylene)s (OPEs) and oligo(phenylenevinylene)s (OPVs), as well as OPE- and OPV-based stars and dendrimers,[38d,40] necessitating development of efficient routes into such species.[41] Indeed, in accessing ruthenium alkynyl-based dendrimers, a facile synthesis of dendrons by ‘steric control’ was developed. This exploits the size of the trans-[RuCl(X)(dppe)2] unit to ensure its reaction with 1,3,5-triethynylbenzene proceeds at two of the three ethynyl units only. The reaction thereby generates a ‘wedge’ for dendrimer construction in one step and without the need for classical organic protection–deprotection sequences (Scheme 1). Studies of this broad range of ruthenium alkynyl-based complexes have revealed that ferrocenyl- and OPE-linked diruthenium complexes exhibit some of the largest cubic non-linearities seen for organometallics thus far,[42] while a new type of dendritic effect, namely a non-linear increase in non-linearity with increase in dendrimer generation, has been identified with ruthenium alkynyl-based dendrimers.[40i,43] Analysis of electroabsorption data for such complexes has revealed that the important excited states responsible for NLO properties are ‘arm-localized’ rather than globally delocalized with octupolar or dendritic complexes.[36c,37,43] A key concern is that these NLO effects are wavelength-dependent, and so there is a need to obtain the spectral dependence of the cubic NLO coefficients (indeed, the first examples of such studies for inorganic complexes were of ruthenium alkynyl complexes); however, it is still the case that data are not routinely available as spectral dependence studies, and this has stymied the development of structure–property relationships.[44]

|
Higher-Order NLO Properties of Metal Alkynyl Complexes
Intensity-dependent absorption has attracted significant attention over the past two decades because materials exhibiting such effects have possible uses in frequency up-conversion lasing, imaging, and microscopy, optical power limiting, microfabrication, data storage and processing, and various biological and medical applications, by exploiting the tight spatial control and the possibility of functioning at longer wavelengths (e.g. in the biological window or in the telecommunications windows). The molecular two-photon absorption cross-section σ2 can be obtained from the absorptive component of the non-linearity and this has been the figure-of-merit employed to compare the performance of disparate molecules. More recently, higher-order multiphoton absorption (three-photon absorption, 3PA, a fifth-order NLO effect, and four-photon absorption, 4PA, a seventh-order NLO effect) has attracted attention because the higher-order intensity dependence promises even greater spatial control of the excitation volume as well as access to effects at longer wavelengths.[45] The Z-scan experiment affords both the non-linear refractive and non-linear absorptive components of the third-order non-linearity, but because it samples the intensity dependence of the non-linearity, it can also afford data corresponding to higher-order NLO effects. Such multiphoton absorption studies are still rare (for example, there have only been ~10 reports of 4PA cross-sections). Studies of ruthenium alkynyl dendrimers have afforded record values of the molecular 3PA cross-section σ3[40l,46] and molecular 4PA cross-section σ4,[46] as well as three-photon absorption-induced photochemistry (Table 3).[40f]

|
Metal Alkynyl Complexes as NLO Switches
Molecular switches, molecules that can be reversibly interconverted between two or more states, have been of long-standing interest (viz. pH indicators), but more recently, the focus has shifted to molecules utilizing other switching modes (in particular, photochromism). The read-out following the switching procedure is often a change in linear optical behaviour (absorbance, fluorescence, etc.) However, it is also possible to use a change in NLO behaviour as the read-out distinguishing the switching states. The early examples of molecular NLO switches have been summarized,[47] with initial studies of organic compounds followed by investigations of coordination complexes and organometallic complexes. The first example of NLO switching in organometallic complexes employed ruthenium alkynyl complexes and protonation–deprotonation protocols (Fig. 7).[32a] Alkynyl complexes have been long known to react with electrophiles and thereby afford vinylidene complexes in a facile fashion,[48] and certain electrophiles (such as [RN2]+ and H+) can be subsequently smoothly removed. With this in mind, the quadratic NLO activity for vinylidene and alkynyl complex pairs was examined, with several examples identified where interconvertible complexes correspond to protically switchable NLO materials. In a similar fashion, the cubic NLO performance of related vinylidene and alkynyl complexes has afforded several examples of protically switchable cubic NLO materials.[32a]

|
Subsequently, attention turned to electrochemical NLO switching, exploiting the reversible redox activity exhibited by many ruthenium alkynyl complexes. Small ruthenium phenylalkynyl complexes have no measurable cubic non-linearity, but on oxidation, an absorption saturation effect is observed; the cubic non-linearity is switched on, the facile reversibility of this process affording an electrochemical cubic non-linearity on/off switch, in the first example of in situ electrochemical switching of optical non-linearity and the first switching of molecular non-linear absorption (Fig. 8).[40a,49] Studies employing the degenerate four-wave mixing technique revealed that these specific NLO effects arise from picosecond time-scale processes (suggesting that applications involving molecular ‘switching’ of such properties can in principle be exceptionally fast).[40b,43]

Switching between three cubic NLO ‘states’ was then demonstrated for the first time, exploiting the widely differing oxidation potentials of the metals in an iron–ruthenium complex,[50] closely followed by ‘orthogonal’ switching of NLO properties across three states for the first time (using protic and electrochemical stimuli) in a ruthenium alkynyl cruciform complex.[51] A specific dithienylperfluorocyclopentene-bridged diruthenium complex was then shown to be able to exist in a record six independently addressable and switchable ‘states’ with distinct linear and NLO properties that can be interconverted by seven distinct pathways exploiting orthogonal protic, electrochemical, and photochemical switching (Fig. 9). Such complexes have the potential to be used in the construction of multi-input logic gates responding to diverse stimuli across a broad spectral range.[52] These new effects are non-linear optical analogues of well-established phenomena (halochromism (acidochromism, protochromism), electrochromism, photochromism) in which linear optical properties are modified on application of an external stimulus, and for which materials are already in commercial use (e.g. pH indicators, smart windows, sunglasses respectively); for the new effects, both non-linear refractive and non-linear absorptive ‘chromisms’ have been demonstrated and therefore have potential application interest.

|
NLO Merit of Metal Alkynyl Complexes Compared with Organic Compounds
Structure–NLO property outcomes have generally been developed from studies by a specific research group using one technique under certain measurement conditions (NLO effects depend on several factors including laser pulse length, pulse repetition rate, laser power, and wavelength) and with certain theory assumptions. However, there is a need to compare NLO measurements for disparate compounds measured in different laboratories using various techniques under different experimental conditions. There are a range of possibilities for scaling NLO data to facilitate comparisons that have been explored,[40l,40m,53] the most widely accepted of which is by molecular weight (owing to ease of implementation) – other scaling factors include molecular volume, number of ‘effective’ (π-delocalizable) electrons, and cost of production. Results from these scaling studies suggest that the NLO efficiencies of ruthenium alkynyl complexes are superior to comparable organics, but data comparisons are still limited and further studies are needed for a definite conclusion.
Conclusion
Ruthenium alkynyl complexes have been shown to exhibit exceptional NLO efficiencies. In several cases, these complexes exhibit world record values of the key NLO coefficients. Not only can they offer superior NLO efficiencies to the best organic materials, they also possess advantages due to their more facile switching between states with differing (positive/negative or on/off, or positive/zero/negative) NLO responses. Propagating these advantages to the solid state and thereby devices is a future challenge, but some reports are now extant, including SHG from ruthenium alkynyl complexes embedded in polymer hosts and oriented in thin films by corona poling,[54] and THG, all-optical switching, and optical data storage applications from ruthenium alkynyl-doped polymers,[55] and with promising results thus far.
Conflicts of interest
The author declares no conflicts of interest.
Acknowledgements
MGH thanks his coworkers and collaborators for fruitful interactions. Generous funding support from the Australian Research Council is gratefully acknowledged.
References
[1] (a) The Principles of Nonlinear Optics (Ed. Y. R. Shen) 1984 (Wiley: New York, NY).(b) The Elements of Nonlinear Optics (Eds P. N. Butcher, D. Cotter) 1990 (Cambridge University Press: New York, NY).
(c) Nonlinear Optics (Ed. R. W. Boyd) 1992 (Academic Press: New York, NY).
(d) Handbook of Nonlinear Optics (Ed. R. L. Sutherland) 1996 (Marcel Dekker: New York, NY).
[2] (a) I. R. Whittall, M. P. Cifuentes, M. J. Costigan, M. G. Humphrey, S. C. Goh, B. W. Skelton, A. H. White, J. Organomet. Chem. 1994, 471, 193.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. P. Cifuentes, J. Driver, M. G. Humphrey, I. Asselberghs, A. Persoons, M. Samoc, B. Luther-Davies, J. Organomet. Chem. 2000, 607, 72.
| Crossref | GoogleScholarGoogle Scholar |
[3] I. R. Whittall, M. G. Humphrey, M. Samoc, B. Luther-Davies, D. C. R. Hockless, J. Organomet. Chem. 1997, 544, 189.
| Crossref | GoogleScholarGoogle Scholar |
[4] K. Clays, A. Persoons, Rev. Sci. Instrum. 1992, 63, 3285.
| Crossref | GoogleScholarGoogle Scholar |
[5] M. Sheik-Bahae, A. A. Said, T. Wei, D. J. Hagan, E. W. van Stryland, IEEE J. Quantum Electron. 1990, 26, 760.
| Crossref | GoogleScholarGoogle Scholar |
[6] See for example: C. C. Frazier, M. A. Harvey, M. P. Cockerham, H. M. Hand, E. A. Chauchard, C. H. Lee, J. Phys. Chem. 1986, 90, 5703.
| Crossref | GoogleScholarGoogle Scholar |
[7] M. L. H. Green, S. R. Marder, M. E. Thompson, J. A. Bandy, D. Bloor, P. V. Kolinsky, R. J. Jones, Nature 1987, 330, 360.
| Crossref | GoogleScholarGoogle Scholar |
[8] (a) H. S. Nalwa, Appl. Organomet. Chem. 1991, 5, 349.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. R. Marder, in Inorganic Materials (Eds D. W. Bruce, D. O’Hare) 1992, pp. 121–169 (Wiley: Chichester, UK)
(c) N. J. Long, Angew. Chem. Int. Ed. Engl. 1995, 34, 21.
| Crossref | GoogleScholarGoogle Scholar |
(d) I. R. Whittall, A. M. McDonagh, M. G. Humphrey, M. Samoc, Adv. Organomet. Chem. 1998, 42, 291.
| Crossref | GoogleScholarGoogle Scholar |
(e) I. R. Whittall, A. M. McDonagh, M. G. Humphrey, M. Samoc, Adv. Organomet. Chem. 1999, 43, 349.
| Crossref | GoogleScholarGoogle Scholar |
(f) M. E. Thompson, P. E. Djurovich, S. Barlow, S. Marder, in Comprehensive Organometallic Chemistry III (Eds R. H. Crabtree, D. M. P. Mingos) 2007, Vol 12, pp. 101–194 (Elsevier: Oxford).
(g) J. P. L. Morrall, G. T. Dalton, M. G. Humphrey, M. Samoc, Adv. Organomet. Chem. 2007, 55, 61.
| Crossref | GoogleScholarGoogle Scholar |
[9] J. C. Calabrese, L.-T. Cheng, J. C. Green, S. R. Marder, W. Tam, J. Am. Chem. Soc. 1991, 113, 7227.
| Crossref | GoogleScholarGoogle Scholar |
[10] S. Barlow, H. E. Bunting, C. Ringham, J. C. Green, G. U. Bublitz, S. G. Boxer, J. W. Perry, S. R. Marder, J. Am. Chem. Soc. 1999, 121, 3715.
| Crossref | GoogleScholarGoogle Scholar |
[11] (a) C. E. Powell, M. G. Humphrey, Coord. Chem. Rev. 2004, 248, 725.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. P. Cifuentes, M. G. Humphrey, J. Organomet. Chem. 2004, 689, 3968.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. A. Green, M. P. Cifuentes, M. Samoc, M. G. Humphrey, Coord. Chem. Rev. 2011, 255, 2025.
| Crossref | GoogleScholarGoogle Scholar |
(d) K. A. Green, M. P. Cifuentes, M. Samoc, M. G. Humphrey, Coord. Chem. Rev. 2011, 255, 2530.
| Crossref | GoogleScholarGoogle Scholar |
(e) G. Grelaud, M. P. Cifuentes, F. Paul, M. G. Humphrey, J. Organomet. Chem. 2014, 751, 181.
| Crossref | GoogleScholarGoogle Scholar |
[12] R. Nast, Z. Naturforsch. B 1953, 8, 381.
| Crossref | GoogleScholarGoogle Scholar |
[13] (a) R. Nast, Coord. Chem. Rev. 1982, 47, 89.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. I. Bruce, M. G. Humphrey, J. G. Matisons, S. K. Roy, A. G. Swincer, Aust. J. Chem. 1984, 37, 1955.
| Crossref | GoogleScholarGoogle Scholar |
(c) I. R. Whittall, M. G. Humphrey, D. C. R. Hockless, Aust. J. Chem. 1997, 50, 991.
| Crossref | GoogleScholarGoogle Scholar |
(d) I. R. Whittall, M. G. Humphrey, D. C. R. Hockless, Aust. J. Chem. 1998, 51, 219.
| Crossref | GoogleScholarGoogle Scholar |
[14] M. I. Bruce, D. N. Duffy, M. G. Humphrey, A. G. Swincer, J. Organomet. Chem. 1985, 282, 383.
| Crossref | GoogleScholarGoogle Scholar |
[15] (a) C. Bruneau, P. H. Dixneuf, Acc. Chem. Res. 1999, 32, 311.
| Crossref | GoogleScholarGoogle Scholar |
(b) B. M. Trost, A. McClory, Chem. Asian J. 2008, 3, 164.
| Crossref | GoogleScholarGoogle Scholar |
[16] J. E. McGrady, T. Lovell, R. Stranger, M. G. Humphrey, Organometallics 1997, 16, 4004.
| Crossref | GoogleScholarGoogle Scholar |
[17] P. J. Low, Dalton Trans. 2005, 2821.
| Crossref | GoogleScholarGoogle Scholar |
[18] G. Wilkinson, Ruthenium. Available at http://www.rsc.org/periodic-table/element/44/ruthenium (accessed 11 June 2018).
[19] (a) See for example: B. J. Coe, J. L. Harries, J. A. Harris, B. S. Brunschwig, P. N. Horton, M. B. Hursthouse, Inorg. Chem. 2006, 45, 11019.
| Crossref | GoogleScholarGoogle Scholar |
(b) B. J. Coe, J. L. Harries, M. Helliwell, B. S. Brunschwig, J. A. Harris, I. Asselberghs, S. T. Hung, K. Clays, N. Horton, M. B. Hursthouse, Inorg. Chem. 2006, 45, 1215.
| Crossref | GoogleScholarGoogle Scholar |
(c) B. J. Coe, J. L. Harries, M. Helliwell, L. A. Jones, I. Asselberghs, K. Clays, B. S. Brunschwig, J. A. Harris, J. Garin, J. Orduna, J. Am. Chem. Soc. 2006, 128, 12192.
| Crossref | GoogleScholarGoogle Scholar |
[20] (a) I. R. Whittall, M. G. Humphrey, S. Houbrechts, A. Persoons, D. C. R. Hockless, Organometallics 1996, 15, 5738.
| Crossref | GoogleScholarGoogle Scholar |
(b) R. H. Naulty, M. P. Cifuentes, M. G. Humphrey, S. Houbrechts, C. Boutton, A. Persoons, G. A. Heath, D. C. R. Hockless, B. Luther-Davies, M. Samoc, J. Chem. Soc., Dalton Trans. 1997, 4167.
| Crossref | GoogleScholarGoogle Scholar |
[21] I. R. Whittall, M. P. Cifuentes, M. G. Humphrey, B. Luther-Davies, M. Samoc, S. Houbrechts, A. Persoons, G. A. Heath, D. Bogsanyi, Organometallics 1997, 16, 2631.
| Crossref | GoogleScholarGoogle Scholar |
[22] (a) I. R. Whittall, M. G. Humphrey, D. C. R. Hockless, B. W. Skelton, A. H. White, Organometallics 1995, 14, 3970.
| Crossref | GoogleScholarGoogle Scholar |
(b) I. R. Whittall, M. G. Humphrey, A. Persoons, S. Houbrechts, Organometallics 1996, 15, 1935.
| Crossref | GoogleScholarGoogle Scholar |
(c) I. R. Whittall, M. P. Cifuentes, M. G. Humphrey, B. Luther-Davies, M. Samoc, S. Houbrechts, A. Persoons, G. A. Heath, D. C. R. Hockless, J. Organomet. Chem. 1997, 549, 127.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. M. McDonagh, N. T. Lucas, M. P. Cifuentes, M. G. Humphrey, S. Houbrechts, A. Persoons, J. Organomet. Chem. 2000, 605, 184.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. K. Hurst, N. T. Lucas, M. G. Humphrey, I. Asselberghs, R. Van Boxel, A. Persoons, Aust. J. Chem. 2001, 54, 447.
| Crossref | GoogleScholarGoogle Scholar |
[23] C. E. Powell, M. P. Cifuentes, A. M. McDonagh, S. Hurst, N. T. Lucas, C. D. Delfs, R. Stranger, M. G. Humphrey, S. Houbrechts, I. Asselberghs, A. Persoons, D. C. R. Hockless, Inorg. Chim. Acta 2003, 352, 9.
| Crossref | GoogleScholarGoogle Scholar |
[24] (a) A. M. McDonagh, I. R. Whittall, M. G. Humphrey, D. C. R. Hockless, B. W. Skelton, A. H. White, J. Organomet. Chem. 1996, 523, 33.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. M. McDonagh, I. R. Whittall, M. G. Humphrey, B. W. Skelton, A. H. White, J. Organomet. Chem. 1996, 519, 229.
| Crossref | GoogleScholarGoogle Scholar |
(c) B. A. Babgi, A. Al-Hindawi, G. J. Moxey, F. I. Abdul Razak, M. P. Cifuentes, E. Kulasekera, R. Stranger, A. Teshome, I. Asselberghs, K. Clays, M. G. Humphrey, J. Organomet. Chem. 2013, 730, 108.
| Crossref | GoogleScholarGoogle Scholar |
(d) E. Kulasekera, S. Petrie, R. Stranger, M. G. Humphrey, Organometallics 2014, 33, 2434.
| Crossref | GoogleScholarGoogle Scholar |
(e) D. Wei, M. S. Kodikara, M. Morshedi, G. J. Moxey, H. Wang, G. Wang, C. Quintana, C. Zhang, R. Stranger, M. P. Cifuentes, M. G. Humphrey, ChemPlusChem 2016, 81, 621.
| Crossref | GoogleScholarGoogle Scholar |
(f) M. S. Kodikara, R. Stranger, M. G. Humphrey, ChemPhysChem 2018, 19, 1537.
| Crossref | GoogleScholarGoogle Scholar |
(g) M. S. Kodikara, R. Stranger, M. G. Humphrey, Coord. Chem. Rev. 2018, in press.
(h) J. Du, M. S. Kodikara, G. J. Moxey, M. Morshedi, A. Barlow, C. Quintana, G. Wang, R. Stranger, C. Zhang, M. P. Cifuentes, M. G. Humphrey, Dalton Trans. 2018, 4560.
| Crossref | GoogleScholarGoogle Scholar |
[25] (a) Organic Materials for Nonlinear Optics (Eds R. A. Hann, D. Bloor) 1989 (Royal Society of Chemistry: London).
(b) Organic Materials for Nonlinear Optics II (Eds R. A. Hann, D. Bloor) 1991 (Royal Society of Chemistry: London).
[26] B. A. Babgi, M. S. Kodikara, M. Morshedi, H. Wang, C. Quintana, T. Schwich, G. J. Moxey, N. Van Steerteghem, K. Clays, R. Stranger, M. P. Cifuentes, M. G. Humphrey, ChemPlusChem 2018, 83, 630.
| Crossref | GoogleScholarGoogle Scholar |
[27] L. K. Myers, C. Langhoff, M. E. Thompson, J. Am. Chem. Soc. 1992, 114, 7560.
| Crossref | GoogleScholarGoogle Scholar |
[28] R. H. Naulty, A. M. McDonagh, I. R. Whittall, M. P. Cifuentes, M. G. Humphrey, S. Houbrechts, J. Maes, A. Persoons, G. A. Heath, D. C. R. Hockless, J. Organomet. Chem. 1998, 563, 137.
| Crossref | GoogleScholarGoogle Scholar |
[29] J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis 2010 (University Science Books: Sausalito, CA).
[30] S. R. Marder, J. E. Sohn, G. D. Stucky, in Materials for Nonlinear Optics: Chemical Perspectives (Eds S. R. Marder, J. E. Sohn, G. D. Stucky) 1991, pp. 2–30 (American Chemical Society: Washington, DC).
[31] A. M. McDonagh, M. P. Cifuentes, N. T. Lucas, M. G. Humphrey, S. Houbrechts, A. Persoons, J. Organomet. Chem. 2000, 605, 193.
| Crossref | GoogleScholarGoogle Scholar |
[32] (a) S. Hurst, M. P. Cifuentes, J. P. L. Morrall, N. T. Lucas, I. R. Whittall, M. G. Humphrey, I. Asselberghs, A. Persoons, M. Samoc, B. Luther-Davies, A. C. Willis, Organometallics 2001, 20, 4664.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. Hurst, N. T. Lucas, M. P. Cifuentes, M. G. Humphrey, M. Samoc, B. Luther-Davies, I. Asselberghs, R. Van Boxel, A. Persoons, J. Organomet. Chem. 2001, 633, 114.
| Crossref | GoogleScholarGoogle Scholar |
[33] (a) T. N. Fondum, K. A. Green, M. D. Randles, M. P. Cifuentes, A. C. Willis, A. Teshome, I. Asselberghs, K. Clays, M. G. Humphrey, J. Organomet. Chem. 2008, 693, 1605.
| Crossref | GoogleScholarGoogle Scholar |
(b) B. Babgi, L. Rigamonti, M. P. Cifuentes, T. C. Corkery, M. D. Randles, T. Schwich, S. Petrie, R. Stranger, A. Teshome, I. Asselberghs, K. Clays, M. Samoc, M. G. Humphrey, J. Am. Chem. Soc. 2009, 131, 10293.
| Crossref | GoogleScholarGoogle Scholar |
(c) L. Rigamonti, B. Babgi, M. P. Cifuentes, R. L. Roberts, S. Petrie, R. Stranger, S. Righetto, A. Teshome, I. Asselberghs, K. Clays, M. G. Humphrey, Inorg. Chem. 2009, 48, 3562.
| Crossref | GoogleScholarGoogle Scholar |
(d) H. Zhang, M. Morshedi, M. S. Kodikara, G. J. Moxey, G. Wang, H. Wang, C. Quintana, R. Stranger, C. Zhang, M. P. Cifuentes, M. G. Humphrey, ChemPlusChem 2016, 81, 613.
| Crossref | GoogleScholarGoogle Scholar |
[34] A. M. McDonagh, M. P. Cifuentes, M. G. Humphrey, S. Houbrechts, J. Maes, A. Persoons, M. Samoc, B. Luther-Davies, J. Organomet. Chem. 2000, 610, 71.
| Crossref | GoogleScholarGoogle Scholar |
[35] (a) I. Ledoux, J. Zyss, J. S. Siegel, J. Brienne, J.-M. Lehn, Chem. Phys. Lett. 1990, 172, 440.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. Joffre, D. Yaron, R. J. Silbey, J. Zyss, J. Chem. Phys. 1992, 97, 5607.
| Crossref | GoogleScholarGoogle Scholar |
(c) V. R. Thalladi, S. Brasselet, D. Bläser, R. Boese, J. Zyss, A. Nangia, G. R. Desiraju, Chem. Commun. 1997, 1841.
| Crossref | GoogleScholarGoogle Scholar |
[36] (a) I. R. Whittall, M. G. Humphrey, S. Houbrechts, J. Maes, A. Persoons, S. Schmid, D. C. R. Hockless, J. Organomet. Chem. 1997, 544, 277.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. M. McDonagh, M. G. Humphrey, M. Samoc, B. Luther-Davies, S. Houbrechts, T. Wada, H. Sasabe, A. Persoons, J. Am. Chem. Soc. 1999, 121, 1405.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. K. Hurst, M. G. Humphrey, T. Isoshima, K. Wostyn, I. Asselberghs, K. Clays, A. Persoons, M. Samoc, B. Luther-Davies, Organometallics 2002, 21, 2024.
| Crossref | GoogleScholarGoogle Scholar |
[37] S. K. Hurst, N. T. Lucas, M. G. Humphrey, T. Isoshima, K. Wostyn, I. Asselberghs, K. Clays, A. Persoons, M. Samoc, B. Luther-Davies, Inorg. Chim. Acta 2003, 350, 62.
| Crossref | GoogleScholarGoogle Scholar |
[38] (a) I. R. Whittall, M. G. Humphrey, M. Samoc, J. Swiatkiewicz, B. Luther-Davies, Organometallics 1995, 14, 5493.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. M. McDonagh, M. P. Cifuentes, I. R. Whittall, M. G. Humphrey, M. Samoc, B. Luther-Davies, D. C. R. Hockless, J. Organomet. Chem. 1996, 526, 99.
| Crossref | GoogleScholarGoogle Scholar |
(c) I. R. Whittall, M. G. Humphrey, M. Samoc, B. Luther-Davies, Angew. Chem. Int. Ed. Engl. 1997, 36, 370.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. M. McDonagh, M. G. Humphrey, M. Samoc, B. Luther-Davies, Organometallics 1999, 18, 5195.
| Crossref | GoogleScholarGoogle Scholar |
[39] (a) S. Drouet, A. Merhi, G. Grelaud, M. P. Cifuentes, M. G. Humphrey, K. Matczyszyn, M. Samoc, L. Toupet, C. O. Paul-Roth, F. Paul, Tetrahedron 2012, 36, 2192.
(b) A. Merhi, G. Grelaud, N. Ripoche, A. Barlow, M. P. Cifuentes, M. G. Humphrey, F. Paul, C. O. Paul-Roth, Polyhedron 2015, 86, 64.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. Merhi, G. Grelaud, K. A. Green, N. H. Minh, M. Reynolds, I. Ledoux, A. Barlow, G. Wang, M. P. Cifuentes, M. G. Humphrey, F. Paul, C. O. Paul-Roth, Dalton Trans. 2015, 7748.
| Crossref | GoogleScholarGoogle Scholar |
(d) F. Malvolti, C. Rouxel, G. Grelaud, L. Toupet, T. Roisnel, A. Barlow, X. Yang, G. Wang, F. I. Abdul Razak, R. Stranger, M. P. Cifuentes, M. G. Humphrey, O. Mongin, M. Blanchard-Desce, C. O. Paul-Roth, F. Paul, Eur. J. Inorg. Chem. 2016, 3868.
| Crossref | GoogleScholarGoogle Scholar |
(e) A. Triadon, G. Grelaud, N. Richy, O. Mongin, G. J. Moxey, I. M. Dixon, X. Yang, G. Wang, A. Barlow, J. Rault-Berthelot, M. P. Cifuentes, M. G. Humphrey, Organometallics 2018, 37, 2245.
[40] (a) M. P. Cifuentes, C. E. Powell, M. G. Humphrey, G. A. Heath, M. Samoc, B. Luther-Davies, J. Phys. Chem. A 2001, 105, 9625.
| Crossref | GoogleScholarGoogle Scholar |
(b) C. E. Powell, M. G. Humphrey, M. P. Cifuentes, J. P. Morrall, M. Samoc, B. Luther-Davies, J. Phys. Chem. A 2003, 107, 11264.
| Crossref | GoogleScholarGoogle Scholar |
(c) C. J. Adams, L. E. Bowen, M. G. Humphrey, J. P. L. Morrall, M. Samoc, L. J. Yellowlees, Dalton Trans. 2004, 4130.
| Crossref | GoogleScholarGoogle Scholar |
(d) J. P. Morrall, M. P. Cifuentes, M. G. Humphrey, R. Kellens, E. Robijns, I. Asselberghs, K. Clays, A. Persoons, M. Samoc, A. C. Willis, Inorg. Chim. Acta 2006, 359, 998.
| Crossref | GoogleScholarGoogle Scholar |
(e) G. T. Dalton, M. P. Cifuentes, L. A. Watson, S. Petrie, R. Stranger, M. Samoc, M. G. Humphrey, Inorg. Chem. 2009, 48, 6534.
| Crossref | GoogleScholarGoogle Scholar |
(f) R. L. Roberts, T. Schwich, T. C. Corkery, M. P. Cifuentes, K. A. Green, J. Farmer, P. J. Low, T. B. Marder, M. Samoc, M. G. Humphrey, Adv. Mater. 2009, 21, 2318.
| Crossref | GoogleScholarGoogle Scholar |
(g) C. J. Jeffery, M. P. Cifuentes, G. T. Dalton, T. C. Corkery, M. D. Randles, A. C. Willis, M. Samoc, M. G. Humphrey, Macromol. Rapid Commun. 2010, 31, 846.
| Crossref | GoogleScholarGoogle Scholar |
(h) M. Samoc, T. C. Corkery, A. M. McDonagh, M. P. Cifuentes, M. G. Humphrey, Aust. J. Chem. 2011, 64, 1269.
| Crossref | GoogleScholarGoogle Scholar |
(i) K. A. Green, P. V. Simpson, T. C. Corkery, M. P. Cifuentes, M. Samoc, M. G. Humphrey, Macromol. Rapid Commun. 2012, 33, 573.
| Crossref | GoogleScholarGoogle Scholar |
(j) H. Zhao, P. V. Simpson, A. Barlow, G. J. Moxey, M. Morshedi, N. Roy, R. Philip, C. Zhang, M. P. Cifuentes, M. G. Humphrey, Chem. – Eur. J. 2015, 21, 11843.
| Crossref | GoogleScholarGoogle Scholar |
(k) Z. Chen, C. J. Jeffery, M. Morshedi, G. J. Moxey, A. Barlow, X. Yang, B. A. Babgi, G. T. Dalton, M. D. Randles, M. K. Smith, C. Zhang, M. Samoc, M. P. Cifuentes, M. G. Humphrey, ChemPlusChem 2015, 80, 1329.
| Crossref | GoogleScholarGoogle Scholar |
(l) B. Gao, L. M. Mazur, M. Morshedi, A. Barlow, H. Wang, C. Quintana, C. Zhang, M. Samoc, M. P. Cifuentes, M. G. Humphrey, Chem. Commun. 2016, 8301.
| Crossref | GoogleScholarGoogle Scholar |
(m) P. V. Simpson, L. A. Watson, A. Barlow, G. Wang, M. P. Cifuentes, M. G. Humphrey, Angew. Chem. Int. Ed. 2016, 55, 2387.
| Crossref | GoogleScholarGoogle Scholar |
(n) T. Schwich, A. Barlow, M. P. Cifuentes, J. Szeremeta, M. Samoc, M. G. Humphrey, Chem. – Eur. J. 2017, 23, 8395.
| Crossref | GoogleScholarGoogle Scholar |
[41] (a) S. K. Hurst, M. P. Cifuentes, M. G. Humphrey, Organometallics 2002, 21, 2353.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. M. McDonagh, C. E. Powell, J. P. Morrall, M. P. Cifuentes, M. G. Humphrey, Organometallics 2003, 22, 1402.
| Crossref | GoogleScholarGoogle Scholar |
(c) C. E. Powell, M. P. Cifuentes, M. G. Humphrey, A. C. Willis, J. P. Morrall, M. Samoc, Polyhedron 2007, 26, 284.
| Crossref | GoogleScholarGoogle Scholar |
(d) C. E. Powell, S. Hurst, J. P. Morrall, M. P. Cifuentes, R. L. Roberts, M. Samoc, M. G. Humphrey, Organometallics 2007, 26, 4456.
| Crossref | GoogleScholarGoogle Scholar |
[42] (a) S. K. Hurst, M. P. Cifuentes, A. M. McDonagh, M. G. Humphrey, M. Samoc, B. Luther-Davies, I. Asselberghs, A. Persoons, J. Organomet. Chem. 2002, 642, 259.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. K. Hurst, M. G. Humphrey, J. P. Morrall, M. P. Cifuentes, M. Samoc, B. Luther-Davies, G. A. Heath, A. C. Willis, J. Organomet. Chem. 2003, 670, 56.
| Crossref | GoogleScholarGoogle Scholar |
[43] M. P. Cifuentes, C. E. Powell, J. P. Morrall, A. M. McDonagh, N. T. Lucas, M. G. Humphrey, M. Samoc, S. Houbrechts, I. Asselberghs, K. Clays, A. Persoons, T. Isoshima, J. Am. Chem. Soc. 2006, 128, 10819.
| Crossref | GoogleScholarGoogle Scholar |
[44] C. E. Powell, J. P. L. Morrall, S. A. Ward, M. P. Cifuentes, E. G. A. Notaras, M. Samoc, M. G. Humphrey, J. Am. Chem. Soc. 2004, 126, 12234.
| Crossref | GoogleScholarGoogle Scholar |
[45] (a) G. S. He, L. S. Tan, Q. Zheng, P. N. Prasad, Chem. Rev. 2008, 108, 1245.
| Crossref | GoogleScholarGoogle Scholar |
(b) H. H. Fan, L. Guo, K. F. Li, M. S. Wong, J. W. Cheah, J. Am. Chem. Soc. 2012, 134, 7297.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Szeremeta, M. Nyk, D. Wawrzynczyk, M. Samoc, Nanoscale 2013, 5, 2388.
| Crossref | GoogleScholarGoogle Scholar |
[46] M. Samoc, J. P. Morrall, G. T. Dalton, M. P. Cifuentes, M. G. Humphrey, Angew. Chem. Int. Ed. 2007, 46, 731.
| Crossref | GoogleScholarGoogle Scholar |
[47] (a) B. J. Coe, Chem. – Eur. J. 1999, 5, 2464.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. A. Delaire, K. Nakatani, Chem. Rev. 2000, 100, 1817.
| Crossref | GoogleScholarGoogle Scholar |
[48] (a) M. I. Bruce, C. Dean, D. N. Duffy, M. G. Humphrey, G. A. Koutsantonis, J. Organomet. Chem. 1985, 295, c40.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. I. Bruce, M. G. Humphrey, G. A. Koutsantonis, B. K. Nicholson, J. Organomet. Chem. 1985, 296, c47.
| Crossref | GoogleScholarGoogle Scholar |
[49] C. E. Powell, M. P. Cifuentes, J. P. L. Morrall, R. Stranger, M. G. Humphrey, M. Samoc, B. Luther-Davies, G. A. Heath, J. Am. Chem. Soc. 2003, 125, 602.
| Crossref | GoogleScholarGoogle Scholar |
[50] (a) M. Samoc, N. Gauthier, M. P. Cifuentes, F. Paul, C. Lapinte, M. G. Humphrey, Angew. Chem. Int. Ed. 2006, 45, 7376.
| Crossref | GoogleScholarGoogle Scholar |
(b) N. Gauthier, C. Olivier, S. Rigaut, D. Touchard, T. Roisnel, M. G. Humphrey, F. Paul, Organometallics 2008, 27, 1063.
| Crossref | GoogleScholarGoogle Scholar |
(c) N. Gauthier, G. Argouarch, F. Paul, M. G. Humphrey, L. Toupet, S. Ababou-Girard, H. Sabbah, P. Hapiot, B. Fabre, Adv. Mater. 2008, 20, 1952.
| Crossref | GoogleScholarGoogle Scholar |
(d) N. Gauthier, G. Argouarch, F. Paul, L. Toupet, A. Ladjarafi, K. Costuas, J.-F. Halet, M. Samoc, M. P. Cifuentes, T. C. Corkery, M. G. Humphrey, Chem. – Eur. J. 2011, 17, 5561.
| Crossref | GoogleScholarGoogle Scholar |
(e) G. Grelaud, N. Gauthier, Y. Luo, F. Paul, B. Fabre, F. Barriere, S. Ababou-Girard, T. Roisnel, M. G. Humphrey, J. Phys. Chem. C 2014, 118, 3680.
| Crossref | GoogleScholarGoogle Scholar |
[51] G. T. Dalton, M. P. Cifuentes, S. Petrie, R. Stranger, M. G. Humphrey, M. Samoc, J. Am. Chem. Soc. 2007, 129, 11882.
| Crossref | GoogleScholarGoogle Scholar |
[52] K. A. Green, M. P. Cifuentes, T. C. Corkery, M. Samoc, M. G. Humphrey, Angew. Chem. Int. Ed. 2009, 48, 7867.
| Crossref | GoogleScholarGoogle Scholar |
[53] T. Schwich, M. P. Cifuentes, P. A. Gugger, M. Samoc, M. G. Humphrey, Adv. Mater. 2011, 23, 1433.
| Crossref | GoogleScholarGoogle Scholar |
[54] (a) A. Migalska-Zalas, B. Sahraoui, I. V. Kityk, S. Tkaczyk, V. Yuvshenko, J.-L. Fillaut, J. Perruchon, T. J. J. Muller, Phys. Rev. B 2005, 71,
| Crossref | GoogleScholarGoogle Scholar |
(b) J. Luc, J. Niziol, M. Sniechowski, J.-L. Fillaut, B. Sahraoui, O. Krupka, Mol. Cryst. Liq. Cryst. 2008, 485, 990.
| Crossref | GoogleScholarGoogle Scholar |
(c) B. Sahraoui, J. Luc, A. Meghea, R. Czaplicki, J.-L. Fillaut, A. Migalska-Zalas, J. Opt. A 2009,
| Crossref | GoogleScholarGoogle Scholar |
[55] (a) J. Luc, J.-L. Fillaut, J. Niziol, B. Sahraoui, J. Optoelectron. Adv. Mater. 2007, 9, 2826.
(b) K. N. Gherab, R. Gatri, J.-L. Fillaut, J. Luc, B. Sahraoui, J. Niziol, Nonlinear Opt. Quantum Opt. 2008, 38, 281.
(c) R. Gatri, J.-L. Fillaut, J. Mysliwiec, A. Szukalski, S. Bartkiewicz, H. El-Ouazzani, I. Guezguez, F. Khammar, B. Sahraoui, Chem. Phys. Lett. 2012, 535, 106.
| Crossref | GoogleScholarGoogle Scholar |
(d) J. Niziol, J.-L. Fillaut, M. Sniechowski, F. Khammar, B. Sahraoui, Opt. Mater. 2012, 34, 1670.
| Crossref | GoogleScholarGoogle Scholar |
(e) J. Luc, K. K. Bouchouit, R. Czaplicki, J.-L. Fillaut, B. Sahraoui, Opt. Express 2008, 16, 15633.
| Crossref | GoogleScholarGoogle Scholar |
(f) K. N. Gherab, R. Gatri, Z. Hank, B. Dick, R.-J. Kutta, R. Winter, J. Luc, B. Sahraoui, J.-L. Fillaut, J. Mater. Chem. 2010, 20, 2858.
| Crossref | GoogleScholarGoogle Scholar |
* Mark G. Humphrey is the recipient of the 2017 RACI Leighton Memorial Medal.