

High Oxidation State Organopalladium(iv) Species: Applications to C–H Activation*,**
Liam K. Burt A B and Curtis C. Ho A B
A B
A School of Natural Sciences – Chemistry, University of Tasmania, Hobart, Tas. 7001, Australia.
B Corresponding authors. Email: lburt1@utas.edu.au; curtis.ho@utas.edu.au
Australian Journal of Chemistry 71(10) 753-755 https://doi.org/10.1071/CH18233
Submitted: 21 May 2018 Accepted: 2 July 2018 Published: 14 August 2018
Abstract
This highlight article focuses on some seminal discoveries in organopalladium(iv) chemistry and the implications and notable applications of these findings in contemporary palladium catalysis and in particular C–H activation.
The development of palladium-catalysed couplings has provided unprecedented access to a diverse range of organic molecules.[1–5] The majority of these processes feature putative palladium(0)/palladium(ii) catalytic cycles. However, initially, considerably less investigation has been conducted with cycles involving palladium(ii)/palladium(iv) redox pairs due to few reported organopalladium(iv) species in early work,[6–9] with unambiguous observation of palladium(iv) limited to perfluorophenyl complexes.[8] Several suggestions as to the possible involvement of organopalladium(iv) species as intermediates were claimed, however, the isolation of hydrocarbylpalladium(iv) complexes was elusive up until 1986. More recently, palladium(iv) has been applied for a range of transformations such as C–H functionalisation and alkene difunctionalisation.[10]
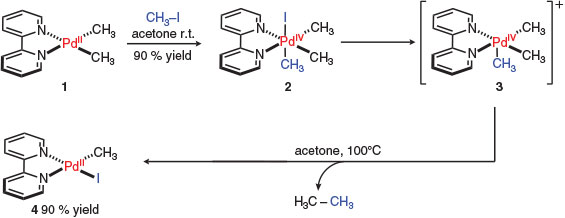
In 1986, Canty, one of the pioneers of high oxidation state organopalladium chemistry,[11,12] reported the first synthesis, isolation, and X-ray crystal structure of a trimethylpalladium(iv) complex.[13–15] This was achieved by the facile oxidative addition of iodomethane to palladium(ii) complex 1 to provide palladium(iv) complex 2 at ambient temperature (Scheme 1). Isolable species 2 was stabilised by a rigid, bidentate bipyridyl ligand and, in contrast, switching to a more flexible chelating ligand such as tetramethylethylenediamine (TMEDA) led to decomposition.[14,16] Monitoring the subsequent reductive elimination by 1H NMR spectroscopy revealed a cationic species (intermediate 3) with all Pd–CH3 bonds remaining intact suggesting that iodide dissociation preceded ethane generation and structure 4 was formed via a cationic pentacoordinate palladium(iv) intermediate before the re-coordination of iodide.[17,18] The use of bipyridyl ligands to stabilise palladium(iv) species was first demonstrated in this study,[13] and represented a significant advance in the area. Indeed, the bipyridyl motif has come to represent a key structural feature of the overwhelming majority of chemistry featuring palladium(iv) species.
|
Canty’s work implied that, relative to palladium(ii) species, palladium(iv) intermediates are usually not prone to facile β-hydride elimination or reduction to palladium(0) and formation of palladium black.[11,14,15,19] These features provided opportunities to complement more conventional catalytic methods featuring lower oxidation state palladium species. However, for palladium(iv) intermediates to find broader applications in catalysis, further research enabling the selective and efficient oxidation of palladium(ii) to palladium(iv) was required.[14,15,19] Cyclic voltammetry studies have been conducted that helped enable the establishment of suitable reagents to effect the oxidation of palladium(ii) to palladium(iv).[20] Hypervalent iodine species were identified as appropriate reagents for the efficient and selective oxidation of palladium(ii), stoichiometrically[21] and catalytically.[12,19] Other investigations involved reacting these mild oxidants with palladium(ii) to promote C–H activation, with a likely mechanism beginning with C–H activation at a palladium(ii) centre followed by oxidation along with further reactivity at a palladium(iv) centre.[22]
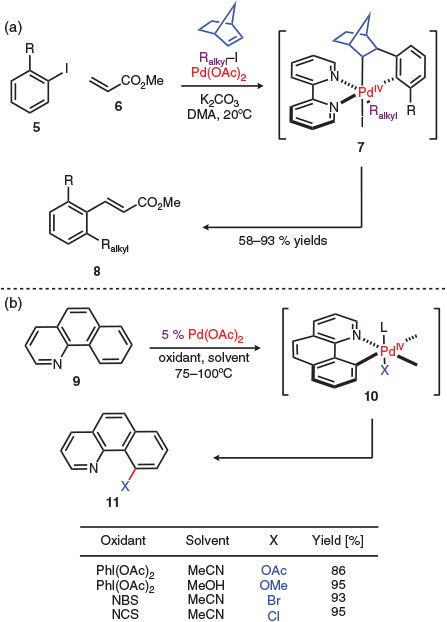
One of the first catalytic reactions invoking a palladium(iv) intermediate was reported by Catellani in 1995 (Scheme 2a).[23,24] This reaction proceeds via C–H activation at a palladium(ii) centre to form a 5-membered high oxidation state palladacycle 7 which ultimately enables the formation of three new C–C bonds in a single step resulting in a Heck-type termination event to yield the functionalised α,β-unsaturated ester (Scheme 2). Similar reactivity allowed for the efficient formation of carbon–heteroatom bonds where the heteroatom originates from the oxidant or solvent (Scheme 2b).[25] In this work, arene C–H activation by a palladium(ii) centre is followed by oxidation to a palladium(iv) intermediate 9 then reductive elimination closes the catalytic cycle. This is the prevailing strategy that has been employed in the development of this class of C–H activation reactions.
|
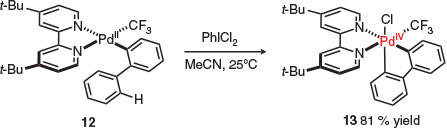
Notably, Sanford and co-workers demonstrated C–H activation at a palladium(iv) centre (i.e. post oxidation) for the first time in 2011 (Scheme 3).[26] This significant study suggests that more complex reaction design and other types of activation steps are possible in higher oxidation palladium catalysis. As C–H activation and reductive elimination are competing pathways, the use of rigid bidentate N-donor ligands (e.g. bipyridine derivatives, substituted N,N′-ethylenediamines, 1,10-phenanthrolines) were crucial to developing an efficient process by inhibiting the reductive elimination and increasing the rate of C–H functionalisation.[12,27–33]
|
Another mechanistic study of these systems conducted by Canty and Sanford found that the isomerisation of the five coordinate cationic palladium(ii) species formed after dissociation of chloride is the rate-determining step and predicted that 5-membered palladacycles are easily accessible, which provided more useful information for the future design of catalytic reactions.[34]
Commencing from original work by Canty and co-workers, palladium(iv) has seen increasing attention for applications in catalysis. Implied through early investigations, Canty suggested that reductive elimination from palladium(iv) complexes is less prone to decomposition relative to palladium(ii) and exploitation of this fundamental reactivity has allowed for the development of palladium(iv)-mediated C–H activation in catalysis.[11,12,35] With the most recent discovery of C–H functionalisation at the metal centre post oxidation, palladium(iv) has been shown as a capable system that can be expanded in many areas in the future.[36]
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
L.K.B. gratefully acknowledges the University of Tasmania for a 2018 Marshall Hughes Honours Scholarship in Chemistry and the Royal Australian Chemical Institute for the 2017 Masson Memorial Scholarship Prize. The authors thank Dr Alex C. Bissember for helpful discussions.
References
[1] R. F. Heck, Ann. N. Y. Acad. Sci. 1977, 295, 201.| Crossref | GoogleScholarGoogle Scholar |
[2] N. Miyaura, A. Suzuki, Chem. Rev. 1995, 95, 2457.
| Crossref | GoogleScholarGoogle Scholar |
[3] R. Chinchilla, C. Nájera, Chem. Rev. 2007, 107, 874.
| Crossref | GoogleScholarGoogle Scholar |
[4] N. T. S. Phan, M. V. D. Sluys, C. W. Jones, Adv. Synth. Catal. 2006, 348, 609.
| Crossref | GoogleScholarGoogle Scholar |
[5] A. B. Kantchev, C. J. O’Brien, M. G. Organ, Angew. Chem. Int. Ed. 2007, 46, 2768.
| Crossref | GoogleScholarGoogle Scholar |
[6] See p. 189 in: A. J. Canty, Handbook of Organopalladlium Chemistry for Organic Synthesis 2002 (John Wiley & Sons: New York, NY).
[7] W. R. Mason, Inorg. Chem. 1973, 12, 20.
| Crossref | GoogleScholarGoogle Scholar |
[8] R. Usón, P. Royo, F. Martinez, J. Organomet. Chem. 1975, 90, 367.
| Crossref | GoogleScholarGoogle Scholar |
[9] D. Milstein, J. K. Stille, J. Am. Chem. Soc. 1979, 101, 4981.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) For a selection of reviews on palladium(iv) in catalysis see: K. Muñiz, Angew. Chem. Int. Ed. 2009, 48, 9412.
| Crossref | GoogleScholarGoogle Scholar |
(b) T. Lyons, M. S. Sanford, Chem. Rev. 2010, 110, 1147.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. J. Hickman, M. S. Sanford, Nature 2012, 484, 177.
| Crossref | GoogleScholarGoogle Scholar |
(d) K. M. Engle, T.-S. Mei, M. Wasa, J.-Q. Yu, Acc. Chem. Res. 2012, 45, 788.
| Crossref | GoogleScholarGoogle Scholar |
(e) J. T. Topczewski, M. S. Sanford, Chem. Sci. 2015, 6, 70.
| Crossref | GoogleScholarGoogle Scholar |
[11] P. Sehnal, R. J. K. Taylor, I. J. S. Fairlamb, Chem. Rev. 2010, 110, 824.
| Crossref | GoogleScholarGoogle Scholar |
[12] K. Muñiz, Angew. Chem. Int. Ed. 2009, 48, 9412.
| Crossref | GoogleScholarGoogle Scholar |
[13] P. K. Byers, A. J. Canty, B. W. Skelton, A. H. White, J. Chem. Soc. Chem. Commun. 1986, 1722.
| Crossref | GoogleScholarGoogle Scholar |
[14] A. J. Canty, Acc. Chem. Res. 1992, 25, 83.
| Crossref | GoogleScholarGoogle Scholar |
[15] A. J. Canty, Dalton Trans. 2009, 10409.
| Crossref | GoogleScholarGoogle Scholar |
[16] W. De Graaf, J. Boersma, W. J. J. Smeets, A. L. Spek, G. van Koten, Organometallics 1989, 8, 2907.
| Crossref | GoogleScholarGoogle Scholar |
[17] P. K. Byers, A. J. Canty, M. Crespo, R. J. Puddephatt, J. D. Scott, Organometallics 1988, 7, 1363.
| Crossref | GoogleScholarGoogle Scholar |
[18] P. K. Byers, A. J. Canty, B. W. Skelton, A. H. White, Organometallics 1990, 9, 826.
| Crossref | GoogleScholarGoogle Scholar |
[19] N. R. Deprez, M. S. Sanford, Inorg. Chem. 2007, 46, 1924.
| Crossref | GoogleScholarGoogle Scholar |
[20] N. M. Camasso, A. J. Canty, A. Ariafard, M. S. Sanford, Organometallics 2017, 36, 4382.
| Crossref | GoogleScholarGoogle Scholar |
[21] A. J. Canty, J. Patel, T. Rodemann, J. H. Ryan, B. W. Skelton, A. H. White, Organometallics 2004, 23, 3466.
| Crossref | GoogleScholarGoogle Scholar |
[22] K. L. Hull, E. L. Lanni, M. S. Sanford, J. Am. Chem. Soc. 2006, 128, 14047.
| Crossref | GoogleScholarGoogle Scholar |
[23] M. Catellani, M. C. Fagnola, Angew. Chem. Int. Ed. Engl. 1995, 33, 2421.
| Crossref | GoogleScholarGoogle Scholar |
[24] M. Catellani, Synlett 2003, 298.
| Crossref | GoogleScholarGoogle Scholar |
[25] A. R. Dick, K. L. Hull, M. S. Sanford, J. Am. Chem. Soc. 2004, 126, 2300.
| Crossref | GoogleScholarGoogle Scholar |
[26] J. M. Racowski, N. D. Ball, M. S. Sanford, J. Am. Chem. Soc. 2011, 133, 18022.
| Crossref | GoogleScholarGoogle Scholar |
[27] A. S. McCall, H. Wang, J. M. Desper, S. Kraft, J. Am. Soc. Chem. 2011, 133, 1832.
[28] J. Vicente, A. Arcas, F. Julia-Hernandez, D. Bautista, Chem. Commun. 2010, 46, 7253.
| Crossref | GoogleScholarGoogle Scholar |
[29] P. L. Alsters, P. F. Engel, M. P. Hogerheide, M. Copijn, A. L. Spek, G. van Koten, Organometallics 1993, 12, 1831.
| Crossref | GoogleScholarGoogle Scholar |
[30] L. R. Gray, D. J. Gulliver, W. Levason, M. Webster, Dalton Trans. 1983, 133.
| Crossref | GoogleScholarGoogle Scholar |
[31] R. Usón, J. Forniés, R. Navarro, J. Organomet. Chem. 1975, 96, 307.
| Crossref | GoogleScholarGoogle Scholar |
[32] N. D. Ball, J. W. Kampf, M. S. Sanford, J. Am. Chem. Soc. 2010, 132, 2878.
| Crossref | GoogleScholarGoogle Scholar |
[33] Y. Ye, N. D. Ball, J. W. Kampf, M. S. Sanford, J. Am. Chem. Soc. 2010, 132, 14682.
| Crossref | GoogleScholarGoogle Scholar |
[34] A. J. Canty, A. Ariafard, B. F. Yates, M. S. Sanford, Organometallics 2015, 34, 1085.
| Crossref | GoogleScholarGoogle Scholar |
[35] L. M. Xu, B.-J. Li, Z. Yang, Z.-J. Shi, Chem. Soc. Rev. 2010, 39, 712.
| Crossref | GoogleScholarGoogle Scholar |
[36] J. J. Topczewski, M. S. Sanford, Chem. Sci. 2015, 6, 70.
| Crossref | GoogleScholarGoogle Scholar |
* Liam. K. Burt was awarded the 2017 Masson Memorial Scholarship.
** This paper is dedicated to Emeritus Distinguished Professor Allan J. Canty on the occasion of his 73rd birthday.