

Living Radical Polymerization by the RAFT Process – A Third Update
Graeme Moad A B , Ezio Rizzardo A and San H. Thang AA CSIRO Materials Science and Engineering, Bag 10, Clayton South, Vic. 3169, Australia.
B Corresponding author. Email: graeme.moad@csiro.au

Graeme Moad obtained his B.Sc. (Hons, First Class) and Ph.D. in organic free radical chemistry from Adelaide University. He joined CSIRO in 1979 and is currently a chief research scientist. Dr Moad is author/co-author of over 150 publications, co-inventor of 33 patent families and co-author of the book The Chemistry of Radical Polymerization. His research interests lie in the fields of polymer design and synthesis. In recognition of his work, Dr Moad was recently awarded the RACI’s Battaerd-Jordan Polymer Medal. Dr Moad is a titular member of the IUPAC Polymer Division and a Fellow of the RACI and the Australian Academy of Science. |

Ezio Rizzardo FRACI, FTSE, FAA, FRS received his Ph.D. from the University of Sydney in 1969 and joined CSIRO in 1976 after post-doctoral research at Rice University, RIMAC, and the Australian National University. His CSIRO research has focused on developing methods for controlling free radical polymerization. He is co-author of some 200 journal papers with over 14,000 citations, and co-inventor on 44 worldwide patents. He has received a number of awards including the RACI Australian Polymer Medal, the CSIRO Chairman’s Gold Medal and an Australian Government Centenary Medal. In 2011, he was the co-recipient of the Prime Minister’s Prize for Science. |

San H. Thang completed his Ph.D. in chemistry at Griffith University in 1987. Currently, he is a chief research scientist at CSIRO Materials Science and Engineering, and an adjunct professor at Monash University. San’s research focuses on the interface between biology and polymer chemistry. He has published over 100 papers which have to date received over 10,000 citations. He is responsible for several key inventions in the area of controlled/living radical polymerization; significantly, he is a co-inventor of the RAFT process. San is a Fellow of the Australian Academy of Technological Science and Engineering, and a Fellow of the Royal Australian Chemical Institute. |
Australian Journal of Chemistry 65(8) 985-1076 https://doi.org/10.1071/CH12295
Submitted: 19 June 2012 Accepted: 25 July 2012 Published: 7 September 2012
Abstract
This paper provides a third update to the review of reversible deactivation radical polymerization (RDRP) achieved with thiocarbonylthio compounds (ZC(=S)SR) by a mechanism of reversible addition-fragmentation chain transfer (RAFT) that was published in June 2005 (Aust. J. Chem. 2005, 58, 379). The first update was published in November 2006 (Aust. J. Chem. 2006, 59, 669) and the second in December 2009 (Aust. J. Chem. 2009, 62, 1402). This review cites over 700 publications that appeared during the period mid 2009 to early 2012 covering various aspects of RAFT polymerization which include reagent synthesis and properties, kinetics and mechanism of polymerization, novel polymer syntheses, and a diverse range of applications. This period has witnessed further significant developments, particularly in the areas of novel RAFT agents, techniques for end-group transformation, the production of micro/nanoparticles and modified surfaces, and biopolymer conjugates both for therapeutic and diagnostic applications.
Introduction
Radical polymerization is one of the most widely used processes for the commercial production of high molar mass polymers.[1,2] The emergence of techniques for implementing reversible deactivation radical polymerization (RDRP), which serve to impart living characteristics to the process, has provided a new set of tools for polymer chemists that allow control over the polymerization process whilst retaining much of the versatility of conventional radical polymerization. It is no longer a formidable task to apply radical polymerization to the synthesis of blocks, stars, or other polymers of complex architecture. New materials with the potential of revolutionizing a large part of the polymer industry continue to appear. The polymerization techniques that are receiving greatest attention are nitroxide-mediated polymerization (NMP),[3–6] atom transfer radical polymerization (ATRP),[7–11] and reversible addition–fragmentation chain transfer (RAFT).
The controversy over the use of the terms ‘living’ and ‘controlled’ in describing processes for radical polymerizations such as ATRP, NMP, or RAFT was addressed by the International Union of Pure and Applied Chemistry (IUPAC).[12] IUPAC recommends that the term living polymerization be confined to refer to ‘a chain polymerization from which irreversible chain transfer and irreversible chain termination (deactivation) are absent’. This effectively precludes use of the adjective ‘living’ in describing processes based on radical polymerization.[13] While it is acceptable to describe radical polymerizations such as ATRP, NMP, or RAFT as controlled polymerization, it is also regarded as inappropriate to use ‘controlled’ in an exclusive sense to define a particular class of polymerization processes since the word has an established, much broader, usage. Use of the terms ‘controlled living’, ‘controlled/living’, ‘pseudo-living’, and ‘quasi-living’ in this context is also discouraged. An IUPAC task group has recommended the use of a new term (controlled) reversible deactivation radical polymerization (RDRP) to describe polymerizations, such as ATRP, NMP or RAFT, which entail equilibria between active and dormant propagating species.[14] This term is not intended to have any connotations as to the fraction of living chains that might be present in a particular polymerization process and does not imply any particular degree of control.
It remains acceptable to use the term ‘living radical polymerization’ to describe a hypothetical process in which termination is indeed absent. It is in this context that we use ‘living radical polymerization’ in the title of this review and the previous articles of this series. We do not intend to imply that termination is absent from any of the polymerizations described herein. Many systems do display the observable characteristics normally associated with living polymerizations and in a few cases, termination, while undeniably present, is not detectable using current techniques.
The increasing importance of RAFT polymerization is illustrated by Fig. 1 which shows the cumulative citations for our first communication on RAFT with thiocarbonylthio compounds,[15] the first RAFT patent,[16] and our previous reviews in the Australian Journal of Chemistry.[17–19] Of course, not all papers on RAFT polymerization cite these sources, nor are all of the papers citing these documents directly relevant to RAFT polymerization. Fig. 2 shows that the remarkable growth in publication of papers covering the various forms of RDRP has continued unabated. The total number of papers that relate to ‘RAFT polymerization’ has increased significantly since mid 2009 with more than 1500 papers being published and approximately one-third of papers on RDRP now pertain to the concept ‘RAFT polymerization’.

|

|
This review is primarily intended to cover the literature on RAFT Polymerization that has appeared since publication of the update published in the Australian Journal of Chemistry in late 2009.[19] We also refer to some earlier papers that were not included in that or the earlier reviews. Work cited in the previous reviews[17–19] is only mentioned again where it is necessary to put the more recent work in context.
The last two years has seen the publication of further general reviews detailing the RAFT process, which include general reviews on RAFT polymerization.[20–25] Reviews devoted to specific areas include those on the kinetics and mechanism of RAFT polymerization,[26,27] RAFT agent design and synthesis,[28] the use of RAFT to probe the kinetics of radical polymerization,[29] microwave-assisted RAFT polymerization,[30,31] RAFT polymerization in microemulsion,[32] end-group removal/transformation,[33–36] the use of RAFT in organic synthesis,[37] the combined use of RAFT polymerization and click chemistry,[38] the synthesis of star polymers and other complex architectures,[39–42] the synergistic use of RAFT polymerization and ATRP,[43,44] the synthesis of self assembling and/or stimuli-responsive polymers,[45–47] and the use of RAFT-synthesized polymers in green chemistry,[48] polymer nanocomposites,[49–51] drug delivery and bioapplications,[41,46,47,52–60] and applications in cosmetics[61] and optoelectronics.[62] The process is also given substantial coverage in most recent reviews that, in part, relate to polymer synthesis, living or controlled polymerization or novel architectures. Some of those that include significant mention of RAFT polymerization include reviews on RDRP,[63] mechanism and reagent design,[11,64,65] click chemistry,[66–72] synthesis of telechelics,[73] the polymerization of carbazole-containing monomers,[74] N-vinyl-1,2,3-triazoles,[75] N-vinyl heterocycles,[76] fluoro-monomers,[77] and glycomonomers,[78–80] synthesis of metallopolymers,[81] conjugated block copolymers,[82] dye-functionalized polymers,[83] stimuli-responsive polymers,[84,85] complex architectures,[86,87] polyolefin blocks,[88] biopolymer–polymer conjugates and bioapplications,[59,89–93] polysaccharide modification,[94,95] polymerization in heterogeneous media,[96,97] microwave-assisted polymerization,[65,98,99] industrial prospects for RDRP,[100,101] and applications in the cosmetics industry.[102]
Mechanism of RAFT
The key feature of the mechanism of RAFT polymerization with thiocarbonylthio compounds, as proposed in our first communication on the subject,[15] is the sequence of addition–fragmentation equilibria shown in Scheme 1. Initiation and radical–radical termination occur as in conventional radical polymerization. In the early stages of the polymerization, addition of a propagating radical (Pn•) to the thiocarbonylthio compound [RSC(Z)=S (1)] followed by fragmentation of the intermediate radical (2) provides a polymeric thiocarbonylthio compound [PnS(Z)C=S (3)] and a new radical (R•). Reaction of this radical (R•) with monomer forms a new propagating radical (Pm•). Rapid equilibrium between the active propagating radicals (Pn• and Pm•) and the dormant polymeric thiocarbonylthio compounds (3) by way of the intermediate 4 provides equal probability for all chains to grow and allows for the production of low dispersity polymers. When the polymerization is complete (or stopped), most of chains are dormant [i.e. PnS(Z)C=S (3)], which possess the thiocarbonylthio end-group, and can be isolated as stable materials.

|
The reactions associated with RAFT equilibria shown in Scheme 1 are in addition to those (i.e. initiation, propagation, irreversible transfer, and termination) that occur during conventional radical polymerization. In an ideal RAFT process, the RAFT agent should behave as an ideal transfer agent. Thus, as is the case with radical polymerization with conventional chain transfer, the kinetics of RAFT polymerization should not be directly affected by the presence of the RAFT agent beyond those affects attributable to the different molar mass and molar mass distribution of the reacting species. Radical–radical termination is not directly suppressed by the RAFT process. Living characteristics are imparted only when the molar mass of the polymer formed is substantially lower than that which would be formed under the same conditions but in the absence of a RAFT agent and is such that the number of polymer molecules with RAFT agent-derived ends far exceeds the number formed as a consequence of termination. Many RAFT polymerizations stray from this ideal.
The initialization process (consumption of the initial RAFT agent) in RAFT polymerization has been studied by 1H NMR spectroscopy for NVP† with a series of xanthates which differ in the ‘R’ group.[103] The selectivity of initialization (and the transfer constant of the RAFT agent) was found to depend strongly on the ‘R’ group. A selective initialization is one in which there is substantial conversion to a single unit adduct before any significant formation of two or higher unit adducts occurs. Xanthates 215, 216, 222 > 224–229 provided (Table 9) selective initialization and were suitable for controlling NVP polymerization with some limitations. Xanthate 221 (R = phenylethyl), which gave a long induction period, and 223 (R = t-butyl), which showed poor selectivity, were not recommended for use with NVP.

|

|

|

|

|

|

|

|

|
RAFT Transfer Constants
The efficiency of the RAFT process is determined by the values of two transfer coefficients, Ctr (= ktr/kp) and C–tr (= k–tr/k–β). The rate coefficient for chain transfer (ktr) for a RAFT agent is given by the Eqn 1. The value of ktr depends on the rate of addition of the propagating radical (Pn•) to the RAFT agent and a partition coefficient (φ) which describes the partitioning of intermediate radical (2) between starting materials and products – refer to Scheme 1:
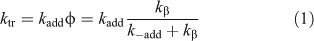
The transfer agent-derived radical (R•) is also partitioned between adding to monomer and reacting with the macro-RAFT agent (3). We therefore define a rate coefficient associated with this reaction (k–tr) as shown in Eqn 2.
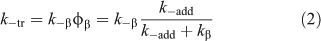
Knowledge of the partition coefficients φ and φβ (note φβ = 1 – φ) and C–tr is important for an understanding of RAFT agent activity. The high reactivity of RAFT agents towards radical addition means that C–tr is seldom zero. Transfer coefficients measured by methods which include an assumption that C–tr is zero or that kβ is zero will typically underestimate Ctr. These values should be called apparent transfer coefficients Ctrapp. In some cases, values of Ctr may be higher than Ctrapp by several orders of magnitude.[104,105] A dependence of Ctrapp on the RAFT agent concentration and/or on monomer conversion is one indication that the reverse reaction is important. The spread in literature values for Ctrapp may be attributed to this effect. Note that the situation is simplified for the case of macro-RAFT agents in homopolymerization where, notwithstanding the effects of chain length, the forward and reverse reactions are the same (Ctr = C–tr) and the partition coefficient φ should be 0.5.
For less active RAFT agents (Ctr ≤ 1), transfer coefficients for RAFT agents may be determined with reasonable accuracy by the usual methods (e.g. the Mayo method). Experimental values of kinetic parameters associated with the RAFT process (addition rate constants (kadd), fragmentation (kβ, k–add) rate constants, and forward (Ctr = ktr/kp), reverse (C–tr = k–tr/ki) and apparent transfer coefficients (Ctrapp)) that have appeared in the literature since, or which were omitted from, our previous review[19] are collected in Table 1.
Gao and Zhu[106] have provided a new analytical expression for estimating values of Ctr of macro-RAFT agents from knowledge of the dispersity and the monomer conversion.
RAFT Equilibrium Constants
The properties of RAFT agents are also often discussed in terms of the value of the equilibrium constants (K = kadd/k–add) associated with radical addition to a thiocarbonylthio compound.[26] Rates of addition are typically high (kadd ~106–108 M–1 s–1). Thus a high equilibrium constant generally implies a low fragmentation rate for the radical adduct and an increased likelihood for retardation and/or side reaction involving this species. In a given RAFT polymerization, there are several equilibrium constants that should be considered.
-
KP (= kaddP/k–addP) associated with the main equilibrium.
-
K (= kadd/k–add) and Kβ = (k–β/kβ) associated with the pre-equilibrium.
-
KR (= kaddR/k–addR) associated with the reaction of the expelled radical with the initial RAFT agent (Scheme 2). Note this is the same as KP when R• is a propagating radical.

|
There are other equilibrium constants to consider if penultimate group effects and the chain length dependence of the various rate constants are considered. Recent reports of values for rate constants for addition (kadd) and fragmentation (k–add) and of the equilibrium constant K are collected in Table 2.
Theoretical calculations have been performed to rationalize the reactivity of RAFT agents and predict RAFT equilibrium and rate constants.[26,116–118] Rodríguez-Sanchez et al.[116,119] used DFT methods and frontier molecular orbital theory to predict the dependence of reactivity of RAFT agents on the ‘R’[119] and ‘Z’ substituents.[116] However, the results are at odds both with experiment and previous theoretical calculations using ab initio or other methods. For example, they[116] suggest an order of reactivity decreasing in the series where Z is Ph > PhCH2 > dithiocarbamate > xanthate > trithiocarbonate. DFT methods were also applied to calculate the molecular structure of dibenzyl trithiocarbonate, and predict its infrared (IR) and ultraviolet (UV-vis) spectra, dipole moment, electrical polarizability, and static first hyperpolarizability.[120]
Lin and Coote[117] indicate that ab initio methods should provide good (within an order of magnitude) prediction of the RAFT equilibrium constant (Keq) and have used these method to estimate that between trithiocarbonate 84 and a (model) poly(MA) propagating radical. Lin and Coote[121] also found that Keq was strongly chain length dependent.
Mechanisms for Retardation
Although the basic mechanism shown in Scheme 1 is generally not disputed, debate continues on the detailed kinetics of the RAFT process, the rapidity with which the various equilibria are established, and what side reactions might occur to complicate the process in specific circumstances.[26,122] In particular the mechanism(s) for retardation in RAFT polymerization mediated by, in particular, dithiobenzoate RAFT agents continue to attract significant interest. The incompatibilities of two mechanistic schemes, namely the slow fragmentation model and the intermediate radical termination model were highlighted in a ‘dilemma paper’ by a IUPAC task group in 2006.[126] The slow fragmentation model points to high concentrations of the intermediate radicals 2 and 4, which are not seen by EPR. The intermediate radical termination model suggests the formation of star polymer byproducts formed by self reaction of 4 or by reaction of 4 with propagating species Pn•, which are not observed in the product in the predicted amounts.
Klumpermann and Heuts[26] reexamined the previously reported calculations of Coote and co-workers and experimental determinations of rate parameters associated with RAFT polymerization using dithiobenzoate agents. They concluded that while the then available data did not allow model discrimination between the schemes, the apparent incompatibility of the models, while significant, was less than suggested in some papers. They,[26] and more recently, Junkers[27] have pointed out the need for more reliable measurements of kinetic parameters to fully resolve the situation.
It has been pointed out that chromatography and molecular weight distributions should distinguish between the slow fragmentation and intermediate radical termination models.[127]
Two theories have been put forward to explain the absence of the star polymer by-products expected if the intermediate radical termination model applies that was mentioned in the previous update. These involve the occurrence of (a) a missing step termination or (b) short chain termination.[128] Ting et al.[129] have attempted to answer the question of whether cross-termination (between 4 and Pn•) might involve short radicals only. They[129] compared the rates of polymerization when a macro-azo-initiator (5) and a polystyrene macro-RAFT agent (6) were used in a RAFT polymerization of styrene (St) (this system should involve no short chain radicals) to the situation where cumyl dithiobenzoate (11) was the RAFT agent and azoisobutyronitrile (AIBN) was used as initiator. Other permutations were also examined. While little retardation was observed when the combination of 5 and 6 (Chart 1) was used, neither was retardation observed with 11 as RAFT agent and AIBN as initiator for higher monomer conversions (>40 %). The latter finding is inconsistent with the short chain termination model of Konkolewicz et al.[128]

|
In very recent work, Meiser and Buback[130] isolated a missing-step product from the reaction of 2-cyanoprop-2-yl radicals with 2-cyanoprop-2-yl dithiobenzoate (12) thereby confirming the viability of this mechanism.
Chernikova et al.[118] used an EPR spin trapping method to measure addition and fragmentation rate constants for the reaction between a t-butyl radical and t-butyl dithiobenzoate (38). Their data analysis indicates a high value for K of ~108 M–1 (20°C). Meiser and Buback[123] used an EPR single pulse method to determine addition and fragmentation rate constants for the reaction between 2-cyanoprop-2-yl radicals radical and 2-cyanoprop-2-yl dithiobenzoate (12). Their analysis indicates a low value for K of ~101 M–1 (60°C). As pointed out by Junkers et al.[131] the more than six orders of magnitude difference in K for these two systems appears surprising. It should, however, be pointed out that significantly different K values for the t-butyl and 2-cyanoprop-2-yl systems might be anticipated since it was already known that 12 and 38 have very different activity as RAFT agents. For example in MMA polymerization (60°C) the values of Ctr for 12 is ~25, and that for 38 should differ little from the value of Ctrapp 0.03;[105] a difference of 103. This, however, can only be attributed in part to a difference in kadd, it is also determined by the partition coefficient φ and the relative rates of fragmentation (k–add) with the two RAFT agents. Chernikova et al.[118] determined kadd to be ~106 M–1 s–1. In their analysis, Meiser and Buback[123] assumed kadd to be ~106 M–1 s–1. It seems unlikely (even allowing for a temperature effect) that the kadd should be the same for the two radicals.
Moad et al. used 13C NMR spectroscopy to follow the initiation of St polymerization with AIBN-α-13C and with cumyl (11), 2-cyanoprop-2-yl (15), and benzyl dithiobenzoates (54) and with cyanoisopropyl dodecyl trithiocarbonate (95).[132] It was found that:
-
Rates of polymerization with dithiobenzoate RAFT agents are strongly dependent on ‘R’ and increase in the series where R is cumyl (11) < 2-cyanoprop-2-yl (15) < benzyl (54).
-
The RAFT agent does not affect the efficiency of AIBN initiation. The rate of formation of St adducts was the same for all RAFT agents.
-
The ketenimine by-product from AIBN is converted into a stable by-product in the presence of RAFT agent. This side reaction will cause some apparent retardation since the ketenimine would normally revert to 2-cyanoprop-2-yl radicals.
-
Some evidence for the missing step termination was observed with benzyl dithiobenzoate (54) as RAFT agent was observed but not with other dithiobenzoates (11 or 15).
Suzuki et al.[133] measured rates of dithiobenzoate-mediated St polymerization in bulk and in miniemulsion. They reported that the significantly higher rates for miniemulsion polymerization could not be simulated with the slow-fragmentation model, but could be adequately understood using the intermediate termination model assuming a relatively high fragmentation rate coefficient and a high rate coefficient for termination between the intermediate radical and a propagating radical.
Some comment should also be made on apparent rate constants for intermediate radical termination in solution. In the studies mentioned above, Chernikova et al.[118] determined kt (intermediate radical termination) to be 6.5 × 102 M–1 s–1 (20°C). Meiser et al.[122] estimated kt (intermediate radical termination) as 0.25 kt (BA polymerization, 60°C).
In RAFT polymerization of St with trithiocarbonate 95, Houshyar et al.[109] found that kt (intermediate radical termination) must be <106 M–1 s–1 (it could be zero). Brown et al.[134] studied RAFT polymerization of St with a dithiocarbamate 254 which contains a potentially florescent carbazole chromophore to aid the detection of star products from intermediate radical termination; no stars were detected.
Further analysis is beyond the scope of this update. Suffice to say that this section does not yet have a conclusion. The mechanisms for retardation in RAFT polymerization, in systems where this is found, is not yet fully resolved.
Macromonomer RAFT Polymerization
Historically, the first RAFT process used to provide living characteristics to radical polymerization was that involving so-called macromonomers (7, 8, Scheme 3).[135–137] Macromonomer RAFT polymerization has been reviewed within larger reviews on catalytic chain transfer (CCT).[138,139] Similarities between the chemistry of RAFT polymerization and that seen in formation and reaction of acrylate mid chain radicals were highlighted in a recent publication.[140]

|
A MAA macromonomer prepared by catalytic chain transfer was used to prepare poly(MAA)-b-poly(BA) which was used as a reactive surfactant in emulsion polymerization of poly(MMA-co-BA-co-MAA).[141] Two papers have appeared providing a comparison of block copolymers synthesized by macromonomer RAFT polymerization with polymers of similar overall composition but synthesized by group transfer polymerization (GTP).[142,143] The copolymers synthesized by GTP had substantially better dispersity (Đ = 1.28) and a more homogeneous composition than those synthesized by macromonomer RAFT (Đ = 1.7) and their superior performance in coatings applications was considered to be a consequence of this.
RAFT-Related Processes
Cobalt-Mediated Polymerization
Polymerizations of VAc and similar monomers mediated by cobalt complexes are proposed to involve a RAFT-like mechanism called associative–degenerative chain transfer (DT) or a reversible coupling mechanism, analogous to that of NMP, called organometallic radical polymerization (OMRP).[144] The relative importance of these mechanisms depend on the specific complex, the reaction conditions, and the monomers. A recent study by Kumar et al.[145] indicates that both the DT and OMRP mechanisms operate simultaneously in the case of VAc polymerization mediated by Co(tmhd)2 (9) (Chart 2).

|
Control in BA polymerization or BA/VAc copolymerization was achieved with use of an alkylcobalt(iii)(acac)2 adduct.[146] Block copolymers of poly(VAc)-b-poly(VPv) or poly(VAc)-b-poly(VBz) were achieved by sequential monomer addition.[147] Poly(VAc) formed by cobalt-mediated polymerization with Co(acac)2 (10) (Chart 2) has been transformed to a polymer with a dithiobenzoate end-group to be used in RAFT polymerization of MAMs.[148]
Polymerization Mediated by Organotellurium, Organobismuthine, and Organostibine Reagents
The mechanism and application of radical polymerization mediated by organotellurium (TERP), organobismuthine, and organostibine compounds has been reviewed.[149,150] Recent papers have described the use of TERP in surfactant-free emulsion polymerization of BA[151] and St,[152,153] the preparation of polymer monoliths based on crosslinked poly(Am),[154] the use of poly(NIPAm)-b-poly(NVP) for solubilization of C60,[155] the use of heteroatom–metal exchange reactions in end-group transformation of polymers (poly(BA), poly(MMA), poly(HEMA), poly(NIPAm)) in organostibine or organobismuth-mediated polymerization[156] and the use of organostibine-mediated polymerization in St/MMA copolymerization.[157]
The end-groups of methacrylic polymers (poly(BA), poly(MMA), poly(HEMA), poly(MAN)) formed by TERP or organostibine-mediated polymerization can be eliminated to form macromonomers in high yield through reaction with (2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) (and formation of an alkoxyamine intermediate) or the addition–fragmentation chain transfer agent, ethyl 2-[(tributylstannyl)methyl]acrylate.[158]
Choice of RAFT Agents
The range of thiocarbonylthio RAFT agents (ZC(=S)SR, 1) continues to expand. The factors which influence choice of RAFT agent for a particular polymerization has been presented in various reviews.[17–19,28,137] The effectiveness of the RAFT agent depends on the monomer being polymerized and is determined by the properties of the free radical leaving group R and the group Z which can be chosen to activate or deactivate the thiocarbonyl double bond of the RAFT agent (1) and modify the stability of the intermediate radicals (2) and (4). For effective RAFT polymerization (Scheme 1, Fig. 3):

|
-
The initial RAFT agents (1) and the polymer RAFT agent (3) should have a reactive C=S double bond (high kadd).
-
The intermediate radicals (2) and (4) should fragment rapidly (high kβ, weak S–R bond in intermediate) and give no side reactions.
-
The intermediate (2) should partition in favour of products (kβ ≥ k–add).
-
The expelled radicals (R•) must efficiently re-initiate polymerization (ki > kp).
A summary of ‘new’ RAFT agents and polymerizations in which they and pre-existing RAFT agents have been applied is provided in Tables 3–14. These tables include some RAFT agent/monomer combinations which provide poorer molar mass control and/or Đ > 1.4. Generally this is indicated by the monomer appearing in parentheses. They are included because they help provide understanding of the mechanism and the construction of guidelines for the choice of RAFT agent. In some cases, poor control may reflect an inappropriate choice of RAFT agent for the monomer or unsuitable reaction conditions. In the tables, the RAFT agents generally appear in order of decreasing homolytic leaving group ability of R. Within a class (e.g. with R = tertiary cyanoalkyl), they generally appear in order of increasing complexity of R. Similarly monomers appear in order of decreasing homolytic leaving group ability of the propagating species (i.e. MAMs > LAMs and methacrylates > methacrylamides > styrenics > acrylates > acrylamides > vinyl monomers).

|

|

|

|

|
Several RAFT agents including dithiobenzoates, 12 and 18, the trithiocarbonates, 89, 95, 98, 123, and 171, and the dithiocarbamate, 258, are now commercially available from Sigma–Aldrich[20] or Strem Chemicals.[21] Lubrizol have announced the availability of trithiocarbonate 125 in metric ton quantities.[159,160]
Dithioesters
RAFT polymerizations making use of dithioester RAFT agents are shown in Tables 3–5. Tertiary dithiobenzoates (11–35; Table 3) continue to be popular RAFT agents particularly for synthesizing polymers based on 1,1-disubsituted monomers (namely, methacrylates (e.g. MMA) or methacrylamides (e.g. HPMAm). The corresponding trithiocarbonates (92–120; Table 7) and aromatic and more active forms of the switchable dithiocarbamates (275-H+, 276-H+; Table 13) are less active but also suitable for controlling the polymerization of these monomers. The aromatic dithioesters are more sensitive to nucleophilic attack and more prone to hydrolysis, for example, when employed in aqueous media. They are also more reactive in end-group transformation/removal reactions post RAFT polymerization.
Trithiocarbonates
RAFT polymerizations making use of trithiocarbonate RAFT agents are shown in Tables 6–8. Two classes of trithiocarbonate RAFT agents are distinguished. Symmetrical trithiocarbonates (Table 6) that have two good homolytic leaving groups and non-symmetrical trithiocarbonates (Table 7) that have one good homolytic leaving group and a poor leaving group such as primary alkyl or aryl.[108] The Z-connected bis-trithiocarbonates shown in Table 8 also have two good homolytic leaving groups.
With symmetrical trithiocarbonates having two good homolytic leaving groups, the trithiocarbonate group usually remains in the centre of the polymer structure. However, the major product from the symmetrical trithiocarbonate 81 in MMA polymerization was the mono-macro-RAFT agent.[320] This outcome is attributed to the substantially better leaving group ability of the MMA propagating radical v. the monomeric analogue.
Xanthates (Dithiocarbonates)
RAFT polymerization making use of xanthate (or dithiocarbonate) RAFT agents are shown in Tables 9 and 10. Xanthate RAFT agents are the most popular for controlling the polymerization of vinyl esters (e.g. VAc) and vinyl amides (e.g. NVP).
Dithiocarbamates and Switchable RAFT Agents
RAFT polymerization making use of dithiocarbamate RAFT agents are shown in Tables 11–13. Cyanomethyl N-methyl-N-phenyldithiocarbamate (258) is commercially available from Aldrich[20] and Strem.[21] In recent work, it has been used for controlling the polymerization of VAc[366] and VC.[529]
Further details of RAFT polymerization using ‘switchable’ RAFT agents that can be switched to offer good control over polymerization of both MAMs and LAMs and a route to polyMAM-b-polyLAM has been reported.[530–532] N-(4-Pyridinyl)-N-methyldithiocarbamates (275–279) are effective with LAMs and the corresponding N-(4-pyridinium)-N-methyldithiocarbamates (275-H+–279-H+) provide excellent control over the polymerization of MAMs (Table 13). In applying the RAFT agents in preparing polyMAM-b-polyLAM, the block comprising MAMs needs to be synthesized first. This sequence is necessary because, polyLAM• are poor homolytic leaving groups with respect to polyMAM• and, consequently, polyLAM-derived macro-RAFT agents have very low transfer coefficients in MAM polymerization. Attempts to synthesize poly(St)-b-poly(VAc) or other poly(St)-b-poly(LAM) starting from a poly(St) macro-RAFT agent gave no significant yield of polymer for an extended period (>4 h) corresponding to the time needed to convert the initial macro-RAFT agent.[530,531] This can be attributed to the very low rate of poly(St)• addition to VAc, possibly compounded by the presence of trace amounts of St monomer in the poly(St) macro-RAFT agent. A solution to this difficulty lay with forming an intermediate ‘block’ of poly(MA).[530,531]
There has been recent emphasis on RAFT polymerization in aqueous media for a variety of reasons which include: (i) perceived environmental benefits, (ii) control over polymerization of monomers with cationic, anionic, zwitterionic, and neutral polar groups which have limited solubility in organic media, and (iii) the need to perform polymerization under physiological conditions. Switchable RAFT agents have been proven effective in the aqueous solution polymerization of N,N-dimethylacrylamide (DMAm).[532] A quantitative study of the influence of acid type and concentration was performed for the polymerization of DMAm.[532] The conclusion was that best control over MAMs is achieved with stoichiometric amounts of a strong acid (e.g. toluenesulfonic acid). The polymerizations were performed with the tetraethyleneglycol macro-RAFT agent 279 which provided enhanced water solubility for the initial RAFT agent in its neutral (unswitched) form.
Polymerizations of LAMs (specifically NVP) in water with the switchable RAFT agents were complicated by the inherent instability of the NVP-based macro-RAFT agents in aqueous media aggravated by the presence of even trace amounts of acid.[532] The recent work of Destarac and co-workers suggests that this problem might be overcome with room temperature polymerization.[506,507] Block copolymers with LAMs were successfully prepared from the DMAm macro-RAFT agent in organic solution.
The N-aryl-N-(4-pyridinyl) dithiocarbamates (273, 274) are more effective (dispersities are lower) than the analogous N-(4-pyridinyl)-N-methyldithiocarbamates (278) with LAMs (NVC, VAc) in the unswitched (neutral) form and more active with MAMs (MA) in their switched (protonated) form.[115] Activity, as indicated by a higher Ctrapp or a lower Đ for the polymer formed, increases with the electron-withdrawing qualities of Ar–X. Some retardation was also observed with the more active RAFT agents of this class.
Other RAFT Agents
Other RAFT agents, namely those with Z = sulfonyl, phosphonate, or phosphine, are shown in Table 14.
The RAFT agents where Z is a strongly electron withdrawing alkyl or phenylsulfonyl group (280, 281) are prone to undergo direct reaction with (meth)acrylate monomers (BA, MA, tBA, and MMA) under polymerization conditions with consumption of the thiocarbonylthio group and ultimately little control over the polymerization.[546] A hetero-Diels Alder mechanism was suggested. Good control was achieved only for iBoA with 280 where the side reactions with monomer were suppressed by steric factors due to the bulky ester substituent.[546,547]
RAFT Agent Selection
Monomers can be considered as belonging to one of two broad classes. The ‘more-activated’ monomers (MAMs) are those where the double bond is conjugated to an aromatic ring (e.g. St, 4VP) a carbonyl group (e.g. MMA, MA, Am) or a nitrile (e.g. AN). The ‘less activated’ monomers (LAMs) are those where the double bond is adjacent to saturated carbon (e.g. DADMAC), an oxygen or nitrogen lone pair (e.g. VAc, NVP) or the heteroatom of a heteroaromatic ring (NVC).
RAFT agents such as dithioesters (Z = aryl or alkyl) or trithiocarbonates (Z = alkylthio) suitable for controlling polymerization of MAMs, inhibit or retard polymerizations of (LAMs). Similarly RAFT agents suitable for controlling polymerizations of LAMs such as N,N-dialkyl- or N-alkyl-N-aryl dithiocarbamates and xanthates tend to have low transfer constants and are ineffective with MAMs.
Fig. 4, which is based on that in our previous reviews but is updated to include switchable RAFT agents,[17–19,137,550] provides a general summary of how to select the appropriate RAFT agent for particular monomers. Note should be made of the dashed lines in the chart. Although some control might be achieved with these monomer–RAFT agent combinations, the molar mass distribution may be broad or there may be substantial retardation or a prolonged inhibition period. This proviso has been omitted in some representations of this data in the literature. The data is presented in another form in Table 15.[551] There are several examples that have been reported (see Tables above) which fall outside of these guidelines and where good control (i.e. low dispersities, successful block synthesis) has, nonetheless, been achieved.

|

|
Synthesis of RAFT Agents
The methods most commonly exploited for the synthesis of (low molar mass) RAFT agents are listed below. The method of choice is dependent on the structure of the desired RAFT agent, the amount required, the toxicity and ease of handling of reagents, and other factors. We have recently completed a critical review of synthetic methods for RAFT agent synthesis which provides further details of the methods listed below.[28]
-
Reaction of a carbodithioate salt with an alkylating agent. O’Reilly and Hansell[474] have published on the synthesis of multifunctional trithiocarbonates The general conditions involve the use of acetone as solvent and phosphate as base as reported in a previous study from the same group.[552]
-
Reaction of a dithiochloroformate or a thiocarbonyl-bis-imidazole with nucleophiles. The esterification of a thiophenol with benzyl dithiochloroformate (e.g. Scheme 4) was used in the synthesis of a series of phenyl trithiocarbonates.[108] The dithiochloroformate may be replaced with the corresponding imidazolide to avoid use of thiophosgene (Scheme 5).[108]
-
Addition of a dithioacid across an olefinic double bond.
-
Radical-induced decomposition of a bis(thioacyl) disulfide.
-
Sulfuration of a thioloester or other substrate.
-
Radical-induced ester exchange.
-
Transesterification (thiol exchange by reaction of a dithioester with a thiol).
-
Base-catalyzed reaction of an activated halide (benzylic halide) with elemental sulfur.
-
Ketoform reaction (used for the synthesis of carboxy functional RAFT agents).

|

|
Many RAFT agents are derived indirectly by modification of other RAFT agents. Further description of those procedures used for the synthesis of macro-RAFT agents from polymers (and biopolymers) formed by non-RAFT processes appears in the section on block copolymers below.
-
Single monomer unit insertion. Chen et al.[433] have published further examples of the synthesis of RAFT agents (150, 151) or macro-RAFT agents (152, see section on block copolymers below) by insertion of a single monomer unit into an existing RAFT agent. Houshyar et al.[109] examined the scope of RAFT single-unit monomer insertion for sequential insertion of St and NIPAm in detail. Critical factors for success were found to be a high transfer constant for the RAFT agent, such that only one monomer unit was added per activation cycle, and a high rate of addition of the radical (R•) to monomer relative to that for further propagation. Single unit monomer insertion can also be favoured over polymerization by use of equimolar amounts of monomer and RAFT agent. Zard and co-workers have reviewed[37] and reported further applications[492,553] of the use of single unit insertion of non-activated monomers into xanthates in organic synthesis. For example, insertion of N-vinylphthalimide provides a synthetic route to amines.[492]
-
Esterification or amidation (carbodiimide)
-
Esterification (acid chloride)
-
Esterification (Mitsunobu reaction)
-
Esterification (other)
-
Active ester-amine reaction or active ester-thiol reaction
-
1,3-Dipolar cycloaddition
-
Other methods
Examples of these processes are found in Tables 5–14 above.
Characterization of RAFT Agents and RAFT-Synthesized Polymers
Most papers on RAFT polymerization contain information on the characterization of RAFT agents, RAFT-synthesized polymers, and/or the RAFT process. In this section we consider papers where the characterization of RAFT agents and RAFT-synthesized polymers by spectroscopic or chromatographic methods is a primary focus.
The UV-visible spectra of an extensive series of an RAFT agents (ZC(= S)SR) have been examined.[554] The position of the absorbance maxima for the π–π* and n–π* transitions and the molar absorptivities were found to be dependent on both the ‘Z’ and ‘R’ substituents. Thus, changes in UV-visible spectra on conversion of an initial RAFT agent to a macro-RAFT agent need to be considered when using spectrophotometry to follow the course of RAFT polymerization. The crystal structure of the RAFT agent 2-[(dodecylsulfanyl)carbonothioylsulfanyl]propanoic acid (155) has been published.[555]
Two reviews contain significant reference to RAFT synthesized polymers.[556,557] The first details recent applications of mass spectrometry, in particular the matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI) techniques, in polymer chemistry. The other is on application of multidimensional chromatography to polymers.[557] This typically involves some combination of high-performance liquid chromatography (HPLC) to separate a polymer sample according to composition and size-exclusion chromatography (SEC) or gel permeation chromatography (GPC) to separate according to molecular weight.[558]
Two dimensional HPLC/GPC methods have been applied to characterize poly(EHA)-b-poly(MA) formed by RAFT dispersion polymerization,[275] the block copolymers formed by hetero-Diels Alder coupling of RAFT-synthesized poly(iBoA) with ATRP-synthesized poly(MMA),[294] and the block and star-poly(St)-PCL formed by sequential RAFT and ring-opening polymerization.[319]
A combination of LC/MS and LC/FTIR was used to characterize poly(NVP) formed with an acid functional xanthate RAFT agent.[498] The rather poor control observed was attributed[498] to extensive chain transfer to dioxan (solvent). The likely acid-catalyzed degradation of the xanthate chain end was not mentioned.
Toxicity of RAFT-Synthesized Polymers
Many recent papers have explored the potential toxicity of RAFT-synthesized polymers. These polymers appear to be generally well tolerated in biological systems.
-
Pissuwan et al.[559] found that, while poly(HPMAm)-dithiobenzoate showed some toxicity with three cell-lines when used at very high concentrations (1000 μM) but were well tolerated when used at lower concentrations (<200 μM), similar polymers with trithiocarbonate ends were non-toxic under all conditions.
-
poly(DMAEMA)-b-poly(HEMA)-dithiobenzoate were shown to have low cytotoxicity with relation to block copolymers based on polyethyleneimine.[217]
-
poly(DMAEA)-trithiocarbonate was shown to have low cytotoxicity and to degrade under physiological conditions to poly(AA).[444]
Polymerization Kinetics
Kinetic simulation is frequently used as a tool to correlate experimental data with theoretical models. The use of RAFT polymerization and kinetic simulation with Predici have been applied to determine chain length dependent termination rate constants in radical polymerizations using the RAFT-CLD-T method.[29] Kinetic simulation has also been used to study the mechanism of microwave-assisted RAFT polymerization of styrene (see below).[560] Kinetic simulation with a method of moments was used to model the effect of backmixing on the outcome of RAFT polymerization for a plug flow tubular reactor or for multiple continuous stirred tank reactors connected in series.[561]
On-line monitoring of RAFT polymerization has been performed using ACOMP (automatic continuous online monitoring of polymerization reactions).[221,439,562] This technique involved passing a stream from the reaction mixture through a series of detectors including a multi-angle light scattering detector, a differential refractometer, a single capillary viscometer, and a photodiode array UV/visible spectrophotometer to provide on-line data on the variation of molecular weight and copolymer composition with polymerization time. The method has been applied to study the homopolymerization of BA in butyl acetate,[439] the copolymerization of BA with MMA,[562] and the copolymerization of DMAEMA with styrene in DMF[221] all with RAFT agent 155.
Reaction Conditions (Initiator, Temperature, Pressure, Solvent, Lewis Acids)
The general guideline for choosing initiator concentrations for RAFT polymerization is that the mole ratio of RAFT agent to amount of initiator decomposed should be >10 : 1 and be such that the molar mass obtained in a control experiment (i.e. same conditions without RAFT agent) is at least 10-fold higher than the desired molar mass.[17] It must be remembered that for every pair of radicals generated, a pair of radicals will terminate to provide dead polymer impurity.
In some cases, it may be necessary to use a higher than desirable initiator concentration to achieve a more acceptable rate of polymerization. Before taking this step, avoidable causes of retardation need to be considered. These causes include: inappropriate choice of RAFT agent, impurities in the RAFT agent (or other components of the polymerization medium), and ineffective exclusion of air or oxygen.
RAFT polymerizations have been successfully conducted at temperatures ranging from sub-ambient to 140°C (or higher under very high pressure). At lower temperature, transfer constants may be lower and retardation may become more important. At higher temperatures, the RAFT agent may be unstable. The importance of these phenomena depends on the specific RAFT agent and monomer combination.
ATRP-Initiated RAFT Polymerization
Several studies have appeared on the use of initiators normally associated with ATRP (halide or sulfonyl halide plus transition metal catalyst) to initiate RAFT polymerization. The process may use a variety of traditional ATRP or SET-RDRP and have been called SET-RAFT.[346,384] Examples include:
-
the use of a metalloenzyme (laccase) as an catalyst and ethyl bromoisobutyrate as initiator to perform RAFT polymerization of PEGMA in the presence of cyanoisopropyl dithiobenzoate (12).[185]
-
polymerization of MA with cyclic RAFT agent 444 and either ethyl 2-bromopropionate or 2-diethyl, meso-2,5-dibromoadipate as initiator and CuBr/tris[(2-pyridyl)methyl]amine catalyst.[563]
RAFT Agent Stability During RAFT Polymerization
Li et al.[221] have reported that some trithiocarbonate RAFT agents (18, 89, 155) are unstable during polymerization of DMAEA at 80°C in DMF. The RAFT agents were stable during BA and St polymerization under similar conditions and a lower rate of degradation was observed in the case of DMAEA/St copolymerizations. The result is surprising since many other groups have reported no specific difficulties in RAFT polymerization of this or other monomers with tertiary amine functionality.
Oxidation of the RAFT agent/end-group was proposed to account for loss of living character during RAFT polymerization of St with 1-phenylethyl dithiobenzoate in air.[162]
RAFT polymerization is not usually compatible with unprotected primary or secondary amines. Janoschka et al.[209] have reported that RAFT polymerization can be successfully carried out in the presence of an unprotected hindered amine (2,2,6,6-tertramethylpiperidene) functionality in the methacrylate 194. However, the polymerization had to be conducted in aqueous ethanol as solvent (polymerization was not successful in toluene) and with a higher than usual concentration of initiator.
Microwave-Assisted RAFT Polymerization
Three reviews on microwave-assisted polymerization[65,98,99] and two reviews specifically on microwave-assisted RAFT polymerization[30,31] have appeared. The number of papers on RAFT polymerizations accelerated by microwave heating continues to increase. Recent examples include RAFT polymerization of MMA,[564] acrylamides (NIPAm, DMAm) and their block copolymers,[392] vinylsilazane (452),[535] and vinyl esters (VAc, VBz, VPv) and their block copolymers.[523] Each of these studies indicate substantial acceleration of polymerization with respect to similar conventionally heated polymerizations.
Controversy over the extent and mechanism of acceleration of the rate of RAFT polymerization continues. Two groups[560,565] have reported on kinetic-modelling of microwave accelerated RAFT polymerization of St in an attempt to gain further understanding of the process. Both came to the conclusion that there is a ‘microwave effect’. The first study[560] attributed accelerated polymerization to an additional initiation process. The second study[565] discounted this possibility and proposed that the results were consistent with the rate constants for addition to monomer (kp) and RAFT agent (kadd) both being accelerated to the same extent by microwave irradiation.
In two independent studies[366,391] of accelerated RAFT polymerization in conventionally heated continuous-flow tubular reactors (see below), microwave-heated batch RAFT polymerizations were conducted as control experiments. No significant differences in polymerization kinetics were observed. These results suggest there is no ‘microwave effect’ beyond that of rapid heating of the reaction medium.
In their perspective, Kempe et al.[99] conclude with reference to apparent ‘microwave effects’ that ‘most of the differences seem to be ascribable to the application of different equipment’ and that ‘these differences might be due to a non-exact control and measurement of the temperature and inhomogeneous electric fields’. In the absence of other data, we concur with this conclusion.
RAFT in Continuous Flow
Solution RAFT polymerization of MMA, BA, VAc, NIPAm, and DMAm has been performed in tubular reactors.[366,391] It was important to degass the polymerization medium and to use metal (stainless steel) tubing for the process.[366] Under these conditions low dispersity polymer similar to those obtained in batch (microwave heated) experiments were obtained. Oxygen-induced inhibition was observed with the use of standard PFA tubing.[366] A procedure for sequential RAFT polymerization and end-group removal in a single flow process has also been reported.[566]
Models have been developed to describe RAFT polymerization in tubular reactors or a series of continuous stirred tank reactors (CSTRs) or a combination of these. Kinetic simulation was then used to predict residence time distributions and the effect of backmixing on the outcome of the process.[561,567] The effect of backmixing is to produce a higher dispersity and a lower average chain length than that expected based on reagent concentrations and conversion, thus it becomes important to exclude reactor backmixing as much as is possible.[561] Methods have also been developed to allow prediction of the monomer sequence distribution for such reactors.[567]
A droplet microfluidic reactor has been described and used to prepared low dispersity poly(St)-b-poly(MMA).[534]
RAFT Polymerization in Ionic Liquids
Ionic liquids have been reported to significantly enhance both the reaction rate and polymer molecular weight in radical polymerization. These effects have been attributed to a reduction in the termination rate constant. Barth et al.[29] have used SP-PLP-EPR measurements to determine termination rate constants (kt) for MMA-d8 polymerization in 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim]BF4) and 1-ethyl-3-methylimidazolium bis(tri-fluoromethylsulfonyl)imide ([emim]NTf2). Both the average <kt> and the value of kt1,1 for reaction between single unit chains were significantly reduced with respect to values for bulk MMA.
Control of Stereochemistry in RAFT Polymerization
Smith et al.[521] observed a higher than usual tacticity (probability of a racemo dyad r = 100 × [rr + mr/2]/[rr + mr + rr] = 69 ± 8 %) for a low molar mass (Mn 1000) poly(VAc) prepared by RAFT polymerization with xanthate 235 at 50°C. A value of r of 53 % was obtained for higher molar mass (Mn > 1300) poly(VAc) for temperatures in the range 37–60°C.
The tacticity of low molar mass NIPAm (Mn 4000) prepared with trithiocarbonate (79) was found to be significantly more syndiotactic (r = 58.6 %) than NIPAm produced by conventional radical polymerizations (without RAFT agent r = 54.6 %).[321] This was attributed to an effect of the acid functional end-group. The syndiotacticity was further enhanced (r = 61.1 %) for polymerization with 3-methyl-3-pentanol as cosolvent.
In order explore the effects of molecular weight and tacticity on the glass transition temperature, Biswas et al.[316] prepared poly(NIPAm) (Mn in the range 70000–100000) with differing tacticity by RAFT polymerization with dithioester 73 in the presence of yttrium triflate (Y(OTf)3) at 60°C. r was in the range 53 % (no Y(OTf)3) to 12 % (0.266 M Y(OTf)3). Katsumoto et al.[284] prepared poly(DEAm)s with differing tacticity by RAFT polymerization in the presence of Y(OTf)3 or Sc(OTf)3.
A high syndiotacticity was obtained in RAFT polymerization of the donor–acceptor–donor monomer, N-(6-acetamidopyridin-2-yl)acrylamide (359), with either 11 or 93 in the presence of 1-octylthymine at 60°C.[170] The value of r was raised from 44 %, for no 1-octylthymine, to 73 %, with 2 molar equivalents of 1-octylthymine with respect to monomer.
A process for forming stereogradient poly(MMA) has been described.[173] The process involves RAFT copolymerization of MAA with a bulky methacrylate such as TPMMA or 1-phenyldibenzosuberyl methacrylate (293) with 11 in toluene solution. Under these conditions, MAA showed increased reactivity, attributed to hydrogen bonding, and was consumed slightly faster than either TPMMA or 293. The copolymers were converted into homo-poly(MAA) by the acid hydrolysis and then to homo-poly(MMA) by esterification with trimethylsilyldiazomethane. The fraction of isotactic triads in the poly(MAA) changed gradually from mm = 14 % to nearly 100 %. RAFT copolymerization of TPMMA and MAA in 1,4-dioxane resulted in consumption of MAA and the bulky methacrylate at the same rate and an atactic polymer (rr/mr/mm = 38/49/13).
RAFT Polymerization in Heterogeneous Media
Several reviews that relate to the use of RAFT in heterogeneous media have appeared[32,96,97] including a tutorial review on RAFT in microemulsion.[32] Examples of RAFT polymerizations in heterogeneous media are included in Tables 3–14 and are distinguished in the tables by the monomer appearing in italics.
Ab initio RAFT emulsion polymerization has been plagued by problems of retardation, colloidal instability, poorly defined molecular weight distributions and broad, possibly multimodal, particle size distributions. Success was strongly dependent on the specific RAFT agent and the monomers polymerized. This is attributed in part to the slow transportation of the RAFT agent between the droplet phase and the particle phase and in part to the way in which molecular weight distributions evolve during RAFT polymerization. Those RAFT experiments in heterogeneous polymerization that were successful were mostly multistep processes, used amphiphilic macro-RAFT agents, and/or made use of miniemulsion or seeded emulsion techniques.
The use of amphiphilic macro-RAFT agents as stabilizers in ‘surfactantless’ polymerization by emulsion, miniemulsion, dispersion and other methods continues to attract attention. Recent examples include:
-
One pot RAFT dispersion polymerization to form PEO-b-poly(St) vesicles in methanol using PEO-macro-RAFT agent 132,[410] and form poly(GMA)-b-poly(HPMA) in water using a poly(GMA) macro-RAFT agent (GMA units are present as 2,3-dihydroxypropyl methacrylate units).[186]
-
Sequential formation of PEGA/MMA macro-RAFT agent (by solution polymerization) and low dispersity poly(PEGA-co-MMA)-b-poly(St) (by emulsion polymerization) in a one-pot process.[354,360] The same macro-RAFT agent was also used in non-aqueous emulsion or dispersion polymerization of BzMA.[361]
-
Hollow nanoparticles were prepared by miniemulsion polymerization of MMA with DVB crosslinker using a MAA/MMA macro-RAFT agent.[368]
-
Shell-crosslinked fluorinated nanocapsules were prepared by miniemulsion polymerization of HA/DFHA with DVB crosslinker using a MAA/DFHA macro-RAFT agent.[369]
-
Emulsion polymerization of VAc was performed with a xanthate-terminated dextran as RAFT agent.[568]
-
Gradient copolymers of TFEMA and AA were synthesized using an amphiphilic RAFT agent and starved feed addition of TFEMA.[438]
-
Emulsion or dispersion polymerization was carried out in supercritical CO2 of VAc with a PEO macro-RAFT agent (245),[526] of MMA with a PDMS macro-RAFT (148),[431] or of NVP with VAc/VPv[513] macro-RAFT agents agent as stabilizer.
-
See also the section below on Microgels and Nanoparticles and Table 24 for examples of RAFT crosslinking polymerization.
RAFT miniemulsion polymerization of St was performed in supercritical CO2 at 50°C with trithiocarbonate 175, an aqueous phase initiator (VA-044), and the anionic surfactant Dowfax 8390.[458] It was found that the particle size could be tuned by changing the CO2 pressure while keeping the recipe constant, with 6.00, 6.50, and 7.50 MPa, generating number-average particle diameters of 98, 89, and 48 nm, respectively.
It has also been found that, particularly in ab initio processes, improved control and colloidal stability is obtained for monomer RAFT agent combinations which have a lower transfer constant. In this context several groups have studied the kinetics of the RAFT emulsion polymerization of St[495,510] with xanthate RAFT agents. A detailed study of RAFT microemulsion of BA with xanthate RAFT agents has also been performed.[32,500,501] It was found that a simplified RAFT mechanism (kadd = ktr, no intermediate radical termination) allowed the kinetics of the microemulsion polymerization of BA with a xanthate RAFT agent to be quantitatively predicted.[32,500] With this model, the value of kβ was found to be important in determining the monomer conversion. Another study used kinetic simulations of a RAFT microemulsion polymerization to show that retardation and inhibition can be explained through consideration of the exit and re-entry of the R• radicals without invoking ‘slow fragmentation’ or ‘intermediate radical termination’ phenomena.[569] Note, however, that the finding that data can bit fit by a simple model does not prove that model.
RAFT polymerization of DMAm in inverse microemulsion has been described.[326,570] The process has been performed with a series of trithiocarbonate RAFT agents.[326] The relative efficacy of these was attributed to the partition coefficient between the aqueous and oil phase.
Polymer self-assembly may be considered an integral part of the initial phase of heterogeneous polymerization and it is well known that amphiphilic polymers of various architectures undergo spontaneous self-assembly in aqueous solution to provide a wide range of nanostructures, which include spherical, worm-like and rod-like micelles, vesicles, nanotubes, and toroids. Nonetheless, the (RAFT) polymerization induced self-assembly and the factors which control what structure is formed have recently attracted considerable interest with studies on the phenomenon in emulsion polymerization,[354,381,400] dispersion polymerization[187,224,229,353,361,571] and non-aqueous dispersion polymerization.[394,397] Blanazs et al.[187] have constructed a phase diagram for poly(288)–poly(HPMA). Refer also to Table 24 for examples of the synthesis of microgels and core-crosslinked star polymers by RAFT-mediated radical crosslinking polymerization most of which can be considered as dispersion polymerizations.

|

|

|

|

|

|

|

|

|
RAFT Polymer Syntheses
Polymer syntheses by RAFT polymerization are summarized in Tables 3–14. Only systems which require separate comment are mentioned here or in the subsequent sections. Some of the more exotic monomers subjected to RAFT polymerization are included in the tables that follow. They include methacrylates (Fig. 5), acrylates (Fig. 6), methacrylamides (Fig. 7), acrylamides (Fig. 8), styrene derivatives (Fig. 9), and vinyl monomers (Fig. 10). Monomers of the above classes with reactive functionality appear in Figs 11 and 12. There continues to be substantial interest in RAFT polymerization of various biorelated monomers which include those derived from amino acids,[572] such as acrylamides 363–368 (Fig. 8), St derivative (384), and glycomonomers, such as methacrylates 312, 313, and 318–320 (Fig. 5), acrylates 341 and 351, methacrylamides 352–355, acrylamide 372, and St derivative 393. Note that such polymers are frequently also made by modification of polymers with reactive post-RAFT polymerization (e.g. those formed from monomers with active ester groups, Fig. 12).

|

|

|

|

|

|

|

|
Methacrylates
A wide range of methacrylates have been successfully polymerized, which include AAEMA, AEMA, AMA, CMA, BMA, BDSMA, DEGMA, DMAEMA, EGMA, HEMA, HMA, iBMA, LMA, MMA, MPC, PEGMA, PFPMA, TMAEMA, TMAPMA, TPMMA (see Abbreviations), (289–333) given in Fig. 5, and compounds 400, 407, 413, and 430. Low dispersities in RAFT polymerization of methacrylates require the use of a RAFT agent with a high transfer constant. This requires that the ‘Z’ group is suitable for MAMs and that the ‘R’ group is a good leaving group with respect to the methacrylate propagating radical. Suitable RAFT agents include dithiobenzoates, 11, 12, 18, and 40, trithiocarbonates, 95 and 98, and aromatic or switchable dithiocarbamates in their more active form, 275-H+ and 276-H+, and derivatives of these.
The polymers poly(NVC)-b-poly(311) and poly(NVC)-b-poly(314) were apparently prepared making use of a poly(NVC) macro-RAFT agent with xanthate RAFT agent 221 and, for the case of poly(NVC)-b-poly(314), good control (a low dispersity polymer) was reported.[496] However, O-alkyl xanthate RAFT agents do not normally provide good control over the polymerization of methacrylates.[573] Furthermore, the poly(NVC) propagating radical is anticipated to be a poor leaving group with respect to the propagating species formed from either 311 or 314.
Monomer 329 containing a xanthate functionality is included in Fig. 6 because with ‘R’ = primary alkyl that functionality does not function as a RAFT agent. It was used as a precursor to a polythiol.
Acrylates
Acrylates mentioned in this survey include AEP, BA, CPA, DA, DEHEA, DMAEA, EA, EAA, EEA, EHA, iBoA, MA, PA, PEGA, tBA, and (334–351) listed in Fig. 6.
Acrylate esters undergo chain transfer to polymer during polymerization leading to branched and even gelled polymers. It has been reported that the extent of branching is higher for conventional radical polymerization than for RDRP (ATRP, RAFT, and NMP).[443] A qualitative explanation was proposed in terms of the differences in the concentrations of short-chain radicals between RDRP and conventional radical polymerization. Reys and Asua[574] have proposed an alternative explanation in terms of radical life times (the time between chain activation and deactivation) being of the same order of magnitude or shorter than the time required for the conformation change necessary for intramolecular hydrogen atom transfer. A recent paper suggests that conventional chain transfer agents (n-octanethiol)[575] and H-bonding solvents (n-butanol)[576] also reduce the extent of branching in conventional radical polymerization.
Methacrylamides
Many examples are appearing of RAFT (co)polymerization of HPMAm; particularly with respect to various bioapplications.[54] Other examples of methacrylamides monomers recently used in RAFT polymerization are the primary amino-functional monomers, AEMAm and APMAm, DEAPMAm, DMAPMAm, 409 and (352–358) shown in Fig. 7. The choice of RAFT agent for polymerization of methacrylamides is subject to similar constraints as mentioned for the methacrylates. In particular the ‘R’ group should be selected so as to be a good leaving group with respect to the propagating radical. The RAFT agents generally preferred include dithiobenzoate 18 and ethyl trithiocarbonate 97 and derivatives of these.
Acrylamides
Attaining low dispersities with RAFT polymerization of acrylamides requires a RAFT agent suitable for MAMs. Acrylamides include Am, AMPS, BzAm, CHAm, DAAm, DEAm, DMAm, DMAEAm, NIPAm, NAM, OAm (see Abbreviations) and (359–372) shown in Fig. 8.
Styrenics
Styrenic monomers subjected to RAFT polymerization include (373–393) in Fig. 9 plus 399, 404, 405, and 406. Good control over RAFT polymerization of St and derivatives requires a RAFT agent suitable for MAMs. Many of the functional styrenes in Fig. 9 have been used to prepare polymers for optoelectronic applications. The advantage of the styrenic monomers in this context is that the functionality is typically not attached through a potentially labile ester or amide linkage.
Diene Monomers
RAFT polymerization of diene monomers generally requires a more active RAFT agent and generally requires use of higher reaction temperatures to obtain good control and reasonable rate of polymerization (e.g. 125°C). RAFT homopolymerizations of BD and Ip in solution are slow.[437] The RAFT copolymerization of Ip with hydroxy-functional monomers, HEMA, HEA, and 394 (Chart 3), with trithiocarbonate (123) has been explored.[396] Diels Alder reaction of Ip with the (meth)acrylates was observed as a side reaction. The (meth)acrylates are consumed preferentially to Ip. thus their copolymers with Ip have a gradient structure. The copolymer of Ip with 394 had a more random structure.

|
Vinyl Monomers
Vinyl monomers in the present context include the vinyl esters (VAc, VB, VBz), the N-vinyl amides and imides (NVCL, NVPip, NVP, NVPI), the N-vinyl heteroaromatics (Vim, NVC), vinylsulfonates (BES, NES), vinylphosphonic acid (VPA), the haloolefins (VF2), and others in Fig. 10. Most of these monomers are LAMs (except 395) and good control over homopolymerization requires use of a RAFT agent suited for that class of monomer (usually a xanthate or a dithiocarbamate). Note, however, that good control over copolymerization of LAMs with MAMs can be obtained with RAFT agents suited to MAMs (e.g. NVPI/NIPAm with 251[517]).
The polymerization of NVP in aqueous media was reported as problematic due to the sensitivity of the macro-RAFT agent with a terminal NVP unit to hydrolysis. It has recently been shown[506,507] that this problem can be overcome by conducting the polymerization at ambient temperature with redox initiation.
A study on the use of benzyl and t-butyl trithiocarbonate and dithiobenzoate RAFT agents for VAc polymerization has appeared.[274] Intermediate radical termination was found to be a significant cause of retardation with these systems.
Monomers with Reactive Functionality
There is a need for processes for polymer modification post-RAFT polymerization (so-called polymer analogous reactions) that proceed in quantitative yield under mild reaction conditions. Several reviews on combined use of RDRP and ‘click chemistry’ have appeared.[66–72] The ‘clickable’ functionality may be present on the Z or R groups of the RAFT agent (see elsewhere in this review) or in the monomers. These monomers can be incorporated in blocks as a precursor to a polymer brush or copolymerized to provide sites for the attachment of functionality or for crosslinking. Many papers concern the combination of RAFT and azide-alkyne 1,3-dipolar cycloaddition. Some alkyne-functional monomers are listed in Fig. 11. Azide functionality is often incorporated post RAFT polymerization.
‘Active ester’ monomers that have been subject to RAFT polymerization (401–405) are shown in Fig. 12. These active ester groups undergo facile reaction with, in particular, substrates with primary amine functionality. Monomers with protected thiol, hydrazide or amino functionality that have been subjected to RAFT (co)polymerization are shown in Fig. 13.

|
Several recent papers have explored polymerization of monomers containing isocyanate or isothiocyanate functionality (Fig. 14).[192,243,288] RAFT polymerization and the thiocarbonylthio group is compatible with isocyanate functionality. However, some care must be taken in selection of the RAFT agent and other components of the polymerization medium such that they do not also contain other functionality that is inherently reactive (such as carboxy).[243]

|
Monomers 416 and 417 with ATRP initiator functionality include CMS and the monomers shown in Fig. 15.[203,587]

|
Crosslinking Monomers
Crosslinking monomers are used both in the synthesis of microgels and polymer networks (see below). This class of monomer includes DEGDMA, TEGDMA, EGDMA, TEGDA, HDDA, MBAm, DVB, and compounds 418–425 (Fig. 16). Use of the monomers such as 420, with an acetal linkage, and 421–423, with disulfide linkages, results in the formation of degradable crosslinks which can be important in controlled release applications. The crosslinks can also be cleaved to allow polymer analysis. See also the section Polymer Networks below.

|
Cyclopolymerization
Further studies on RAFT cyclopolymerization of DADMAC have been reported. The most recent work used xanthate (229)[113,506] or trithiocarbonate (155).[440] RAFT polymerization with xanthate (229) was controlled although dispersities were broad.[113,506] This was attributed to the low transfer constant of poly(DADMAC) macro-RAFT agent in the DADMAC polymerization (Table 1). With trithiocarbonate (155), while the polymerization showed some living character, conversions were low and dispersities were poor (Đ > 1.5) and the latter increased with monomer conversion.[440] While a trithiocarbonate RAFT agent is likely to have a higher transfer constant in DADMAC polymerization, intermediate radical fragmentation is likely to be slower making retardation more likely.
A 1 : 2 mixture of DVE and MAH with dibenzyl trithiocarbonate was reported to undergo alternating RAFT cyclocopolymerization to provide the polymer 426 as shown in Scheme 6.[341,342] Other studies on RAFT cyclopolymerization are summarized in Fig. 17.

|

|
Ring-Opening Polymerization
RAFT ring-opening polymerization provides a simple method of forming polymers with readily cleavable linkages, i.e. esters, thioesters, and/or disulfides, in the carbon–carbon backbone of a polymer chain.[276] The monomers 433–437 (Fig. 18) were copolymerized with MMA, DMAm, and HEMA.

|
End-Functional Polymers and End-Group Transformations
Processes for thiocarbonylthio end-group removal or transformation post RAFT polymerization continue to attract significant interest. Two reviews focussing on end functional polymer and end-group removal/transformation have appeared within the last two years.[33,34] A wide variety of methods are now available for removing or transforming the thiocarbonylthio groups in RAFT-synthesized polymers (Scheme 7[34]). All have advantages and limitations depending on the intended application. The thiocarbonyl functionality present in RAFT-synthesized polymers, once seen as a limitation to the wide-spread adoption of RAFT polymerization can now be seen as an enabling functionality in addressing the needs of the biomedical, optoelectronic, nanotechnology, and other sectors.

|
Radical-Induced Reduction
Radical-induced reduction allows the thiocarbonylthio group of a RAFT-synthesized polymer to be replaced with a hydrogen atom. Recent examples are shown in Table 16. Selective removal of the dodecyl trithiocarbonate ends from poly(CMS) (with pendant benzylic chloride functionality) was successfully performed with tri-n-butylstannane as the H-donor.[436]
Addition–Fragmentation Coupling
A popular method for end-group removal/transformation involves heating the RAFT-synthesized polymer with a large excess of a radical initiator, most often, an azo-compound. That most commonly used is AIBN. However functional azo-compounds (e.g. 438–440, Chart 4) have also been successfully used. Some recent examples are included in Table 17. The strategy appears generally successful for methacrylic polymers (Table 17). The expected complications from primary radical disproportionation are not reported. However, incomplete end-group removal is often found for acrylic and styrenic polymers.[194] The potential limitations of the technology and their causes are discussed in our recent review.[34]

|
Peroxide initiators (LPO, BPO) have been shown to be generally more effective than azo-initiators (e.g. AIBN) in achieving end-group removal.[593] However, the complication of termination by self reaction of propagating species is more pronounced. Chen et al.[194] have shown that a combination of a peroxide initiator (LPO) and azo-initiator (AIBN) is more effective than either alone (Scheme 8). Since the polymers formed are likely to possess end-groups from the peroxide and the azo-initiator, this strategy is most likely not suitable for preparing polymers with defined end-group functionality. The method has also been used as a degrafting process in silica-supported polymer synthesis.[594]

|
Radical-Induced Oxidation
It has been reported that addition of a very small amount of a CCT agent ([(CH3OH)2Co(dmgBF2)2] : [RAFT agent]0 = 1 : 80000) during the latter stages (~70 % monomer conversion) of a RAFT polymerization of a methacrylate monomer (MMA, BMA) provides complete end-group removal and a high purity low dispersity macromonomer product.[602] Details of the mechanism were not presented, however, a possible mechanism combining elements of those for the RAFT and CCT processes is shown in Scheme 9. The process can be considered as a radical-induced oxidation.

|
Reaction with Nucleophiles (Aminolysis/Hydrolysis/Ionic Reduction)
Conversion of thiocarbonylthio end-groups into thiol groups through reaction with nucleophiles is one of the most common end-group transformations. A list comprising recent examples is provided in Table 18. In order to avoid the complication of disulfide formation the process is commonly done in the presence of a reagent to trap the thiol. A summary of the transformations that have been used is provided below (see section entitled Click Reactions).[34]
It has been reported that the use of hydrazine as nucleophile is advantageous both in giving a very rapid reaction and little disulfide formation even when the process was carried out in air.[375] However, Lee et al.[176] found disulfide formation to be significant during the hydrazinolysis of St/AcS copolymers.
Oxidation
Thiocarbonylthio chain-ends of RAFT-synthesized polymers can be efficiently converted into hydroxy chain ends in high yield by heating a THF solution of the polymer and AIBN in air (to produce a polymer with hydroperoxy ends) and then treating this solution with triphenylphosphine.[182,605] The process was demonstrated for polymers with dithiobenzoate or dithiophenylacetate end-groups and for polymers with trithiocarbonate mid-groups (synthesized with dibenzyl trithiocarbonate) (Table 19).
End-groups of poly(MMA) and poly(St) formed with dithiobenzoate 12 are reported to be completely removed by heating a THF solution of the polymer in air.[180] The process was accelerated by addition of acid. The end-group removal process was proposed to involve hydrolysis of the dithiobenzoate to a thiol end-group, however, the end-groups formed were not fully characterized and it seems more likely that the process is analogous to that seen with AIBN/air in THF.
It has also been proposed that hydrogen peroxide can be used to directly transform RAFT end-groups to hydroxy end-groups.[608] The process was applied to poly(NVP) with xanthate chain ends and poly(St) with dithiobenzoate end-groups and involved heating the polymers with hydrogen peroxide at 60°C. The proposed mechanism involved thermal generation of hydroxyl radicals and an addition-fragmentation-coupling process (see below). However, while hydrogen peroxide may undergo induced decomposition, thermal homolysis requires relatively high temperatures.
A facile process in which alkaline hydrogen peroxide is used to transform dithioesters (ZCS2R) to a mixture of a carboxylic acid (ZCO2H) and a disulfide (RSSR) in very high yield has been reported[609] but has not been applied to polymeric substrates.
The radical-induced (radicals from AIBN) air oxidation of RAFT agents and RAFT-synthesized polymers has been investigated by Li et al.[162] The rate of oxidation was greatest for bulky or electron-deficient ‘R’. For a given ‘R’ group, the ease of oxidation of RAFT agents appeared to have an inverse correlation with the activity of the RAFT agent in polymerization and increased in the series where ‘Z’ is phenyl– < benzyl– (dithioesters) < alkylS– (trithiocarbonates) < alkylO– (xanthates). The main reaction observed for all classes of RAFT agent was replacement of the thiocarbonyl group with a carbonyl group. This reaction accounts for some loss of living character during RAFT polymerization in air.
RAFT-synthesized polymers with xanthate end-groups react with ozone quantitatively to form the corresponding polymer with a thiolocarbonate end-group.[606,607] The mechanism was not fully elucidated but the thiocarbonyl is replaced with a carbonyl and sulfuric acid is formed as a by-product (Scheme 10). The process was claimed to also be applicable to other RAFT end-groups (trithiocarbonates, dithioesters) and a patent was filed on the use of this process.[607]
|
|
Thermolysis
An in depth study of the mechanism of thermal decomposition of RAFT agents in solution has appeared.[610] The thermal stability of dithiobenzoates was found to decrease in the series where R is CH2Ph > poly(St) > CH(CH3)Ph > C(CH3)2C2OC2H5 > C(CH3)2Ph > poly(MMA) > C(CH3)2CN. The thermal stability of RAFT agents with R = phenylethyl decreased in the series where Z is OC2H5 > N(C2H5)2 > SCH(CH3)Ph > Ph > CH2Ph > PhNO2. Some correspondence with activity (Ctrapp value) was noted. For RAFT agents possessing β-hydrogens the mechanism of decomposition was proposed to involve Chugaev elimination.
End-group thermolysis has usually been carried out at temperatures >150°C for relatively short reaction times. It has been reported that thermolysis of poly(MMA)-dithiobenzoate in dimethyl sulfoxide solution is complete after heating at 100°C for 24 h.[183] The unsaturated end-group produced was subsequently transformed by thio-Michael reaction with mono(6-deoxy-6-mercapto)-β-cyclodextrin.
Click Reactions
Use of ‘click chemistry’ in combination with RAFT polymerization continues to attract much attention.[38,66–68] The RAFT process can be used to synthesize polymers with clickable moieties at the chain ends through the use of RAFT agents with appropriate functionality on ‘Z’ or ‘R’.
Some reactions in this category are:
-
Copper-catalyzed azide-alkyne 1,3-dipolar cycloaddition. Recent examples of RAFT agents prepared or used that contain an alkyne functionality include dithiobenzoates 22,[255] 28, and 33,[259] trithiocarbonates 118,[379] 136,[267] and 161,[442] and xanthate (231).[518] Those incorporating azide functionality include trithiocarbonates 102,[371] 137,[371] and 138.[419] Examples of the use of this form of click reaction include the synthesis of poly(NIPAm) stars based on a hexakis(fullerene) core.[267] Pressley et al.[611] have reported on an improved methodology for conducting click reactions to form block and cyclic polymers. The process makes use of commercially available copper nanoparticles and is performed with microwave irradiation. The process was claimed to be insensitive to air.
-
The active ester-amine reaction. RAFT agents with active ester functionality include dithiobenzoate 25[259] and trithiocarbonates 130[386] and 181[466].
-
The thiol-halide reaction. The dibromomaleimide group (e.g. 177) has been shown to react with thiols under mild reaction conditions and does not need to be protected during RAFT polymerization of acrylates and acrylamides.[459] Significant retardation was observed with use of 177 in St polymerization.
The thiocarbonylthio functionality can be transformed to a thiol group through reaction with nucleophiles (see above). A variety of thiol transformation reactions, some of them referred to as ‘click’ processes, can then be used. These include the thiol-ene reaction and other processes as shown in Scheme 11.[34,35] Thiol-click reactions have been reviewed[71] and references to some recent examples of these process are included in Table 18.

|
A detailed study of the thiol-ene based Michael addition reactions has been reported[612] in which various catalysts, primary and tertiary amines, and phosphines, were investigated for the reaction of thiols with dimeric and oligomeric (meth)acrylate macromonomers. While primary and tertiary amines were effective catalysts for the thiol–ene reaction, several hours were required to reach high conversions. The phosphine catalysts, dimethylphenylphosphine (DMPP) and tris-(2-carboxyethyl)phosphine (TCEP), were more efficient. DMPP provided complete conversion within a few minutes under optimized conditions. However, the concentration of DMPP had to be kept at very low levels to avoid the formation of by-products originating from the addition of DMPP to the macromonomer. TCEP was an efficient catalyst for thiol-ene reactions in aqueous media when the pH of the medium was higher than 8.0. Under acidic pH the formation of by-products was observed.
An in-depth study of the thiol-epoxy reaction for end-group modification of poly(DEAm) and poly(St) prepared with a dithiobenzoate RAFT agent (18) has appeared.[193] The use of both base (DBU) and Lewis acid catalysis was studied.
A third class of reaction involves direct modification of the RAFT thiocarbonyl functionality:
The Thiocarbonyl-Hetero-Diels Alder Reaction
RAFT agents and macro-RAFT agents with electron withdrawing ‘Z’ undergo hetero-Diels Alder reactions with suitable dienes (Scheme 12).[268,546,613,614] In recent work the process has been developed as a route to block copolymers,[268,296,297,548,615] graft, star, and network polymers,[309,614] and modified surfaces.[295,547,613] The method is also used in synthetic organic chemistry.[616] The process requires RAFT agents or macro-RAFT agents with an electron withdrawing ‘Z’ group. Suitable ‘Z’ groups include 2-pyridyl (57, 70), phosphonate (282–284), or phenylsulfonyl (280). To achieve acceptable rates for reactions in organic solution, the reaction may be catalyzed by a Bronsted or Lewis acid, for example, trifluoroacetic acid or zinc chloride. However, for reaction in aqueous solution no catalyst is necessary.[297] The reaction is thermally reversible[309,548,617] prompting a proposal that it might be used as a thermally stimulated colour switch[548,617] or be applied in reversible block[298] or network formation.[309]

|
The Thiocarbonyl 1,3-Dipolar Cycloaddition Reaction
It has been found that diazomethane undergoes a facile 1,3-dipolar cycloaddition with dithiobenzoate RAFT agents and with dithiobenzoate end-groups of polymers formed by reversible addition–fragmentation chain transfer (RAFT) polymerization.[181] Thus, 2-cyanoprop-2-yl dithiobenzoate (12), on treatment with diazomethane at room temperature, provided stereoisomeric 1,3-dithiolanes in near quantitative (>95 %) yield. A low molar mass RAFT-synthesized poly(MMA) with dithiobenzoate end-groups underwent similar reaction as indicated by immediate decolourization and a quantitative doubling of molar mass. Higher molar mass poly(MMA) were also rapidly decolourized by diazomethane and provided a product with a bimodal molar mass distribution. Under similar conditions, the trithiocarbonate group does not react with diazomethane. The proposed mechanism is shown in Scheme 13.

|
Other End-Group Modification Processes
Staudinger ligation has been proposed as a method for quantitative end-group modification with possible application in forming bioconjugates.[370] RAFT agents 99–101 and 129 were prepared to test the hypothesis. The phosphine functionality in these RAFT agents did not interfere in the RAFT polymerization. For poly(St) formed with 99, Staudinger ligation appeared slow. For poly(St) formed with 100–129, while Staudinger ligation appeared rapid and quantitative, the sensitivity of the phosphine group to oxidation, even when borane-protected, was found to be a complication.[370]
Statistical/Gradient Copolymers
Due to the effects of compositional drift, most copolymers prepared by RAFT polymerization generally fall under the description of gradient copolymers. Many examples of copolymer synthesis are included in the Tables above.
Block Copolymers
Sequential Monomer Addition
Examples of block copolymer synthesis by sequential monomer addition are included in Tables 3–14. See also the section Choice of RAFT Agents for discussion and results on the importance of choosing which monomer to polymerize first and on the use of switchable RAFT agents in this context.
ATRP-RAFT
Thiocarbonylthio compounds have found use as initiators in ATRP.[44] The first reports involved the use of N,N-dialkyl dithiocarbamate derivatives as ATRP initiators. Dithiocarbamates do not provide effective control over the polymerization of MAMs (e.g. St, MMA, MA, AN) in the RAFT process so the control mechanism is most likely that of ATRP.
Recently, more active RAFT agents such as N,N-diaryldithiocarbamates, 1-pyrrolecarbodithioates, trithiocarbonates,[387] and dithioesters (1-dithionaphthalates[206] and dithiobenzoates[168]) have been used as initiators in these polymerizations.[44] Since these are effective RAFT agents with the monomers used, it is pertinent to question the mechanism of the process. While one should not exclude ATRP participation, it seems best to consider these processes as RAFT polymerizations with ATRP initiation. The combination of zero-valent iron (iron powder) and dithioester 16 was used to initiate RAFT polymerization and this may involve a similar mechanism.[205]
Further examples of the synthesis of polymers by a combination of ATRP and RAFT have been reported.[185,525,618] This can involve preparing one block comprising a MAM by ATRP and then a second block comprising a LAM by RAFT.
Matyjaszewski and co-workers have used compounds that combine RAFT agent and ATRP initiator functionality in the one molecule in the synthesis of poly(NVP) blocks (Scheme 14).[525] Bromoisobutyrate derivatives (e.g. 241, successful with VAc) could not be used as RAFT agents in this context because of dimerization of NVP which was catalyzed by trace amounts of HBr.[525] This problem was overcome with the use of the corresponding chloroisobutyrate (242).

|
Further examples of coupling of RAFT-synthesized polymers with ATRP-synthesized polymers by copper-catalyzed click reaction have also been reported.[416]
NMP-RAFT
RAFT end-group transformation by addition–fragmentation-coupling can be performed in the presence of a nitroxide to yield an alkoxyamine chain end.[619] This enables macro-RAFT agents to be transformed to alkoxyamines for use in NMP. Favier et al.[619] developed such a process for exchanging the thiocarbonyl group (ZC(=S)S–) of macro-RAFT agents with a nitroxide (R′2NO–) (Scheme 15), which they termed ESARA; an acronym for ‘exchange of substituents between (macro)alkoxyamines and (macro)RAFT agents’. The process was demonstrated for low molar mass RAFT agents or macro-RAFT agents and SG1-based alkoxyamines.

|
The dithiobenzoates RAFT agents 45 and 46 are also TEMPO-based alkoxyamine initiators for NMP.[280] These agents provided reasonable control over polymerization of St under RAFT conditions (60°C, AIBN initiator) for low monomer conversions although dispersities broadened for higher monomer conversions. Poor control was observed in St polymerization under ‘NMP conditions’ (120°C). Good control (Đ < 1.3) was observed for PEGMA and NIPAm under RAFT conditions but poor control (Đ > 1.7) for AA and tBA under RAFT conditions. Block copolymer formation was also examined.
ROP-RAFT
Hydroxy-end functional polymers prepared by RAFT polymerization can be used as initiators in ring-opening polymerization of cyclic esters (e.g. caprolactone, lactide). Three different strategies have recently been reported. These involve:
-
the use of hydroxy end-functional poly(NIPAm/DMAm) prepared with RAFT agent 37 to prepare low dispersity poly(NIPAm/DMAm)-block-PLA.[270–272]
-
the use of trithiocarbonate 140 to carry out simultaneous RAFT polymerization of St and ROP of lactide.[421] The product was used as a macromonomer in ROMP.
-
transformation of the end groups of poly(St) prepared with phenyldithioacetates 77 or 78 to hydroxy groups by oxidation. Thsese polymers were then used to initiated ROP of caprolactone.[319]
Macro-RAFT Agent Synthesis
Block copolymers based on polymers formed by other polymerization mechanisms are often made by first preparing an end functional pre-polymer which is converted into a macro-RAFT agent by end-group transformation. This macro-RAFT agent is then used in the preparation of the desired block copolymer. Some examples of macro-RAFT agents are shown Tables 3–14 and include PEO macro-RAFT agents (21, 119, 120, 132, 169, 245), PCL macro RAFT agents (66, 225), a PLA macro-RAFT agent (133), polybenzamide macro-RAFT agents (59, 60), polyisoprene macro-RAFT agents (143, 200), a PDMS macro-RAFT agent (148), and poly(3-hexylthiophene) macro-RAFT agents (75, 116, 152, 184, 185). Other examples are mentioned in Table 20. The use of protein-derived macro-RAFT agents has been reviewed.[60]
Esterification/Amidation
A frequently used approach involves esterification or amidation of a substrate possessing amino- or hydroxy-functionality with a carboxy-functional RAFT agent (e.g. Scheme 16). Examples of this strategy are summarized in Table 20.

|
Single Unit Monomer Insertion
Another strategy for preparing macro-RAFT agents involves insertion of a macromonomer unit into a small RAFT agent. The strategy involves careful selection of monomer or reaction conditions such that there is no oligomerization. The strategy has been applied to form poly(3-hexylthiophene) macro-RAFT agent 152.[62,433]
Halogen Substitution
The hydroxyl end-group of PCL formed by ring-opening polymerization was transformed to a xanthate chain end by sequential reaction with 2-bromopropionyl bromide and potassium xanthate, and the resultant macro-RAFT agent used in RAFT polymerization of NVP.[497] For other examples of forming macro-RAFT agents by halogen substitution see ATRP-RAFT above.
Click Reactions
RAFT agents with functionality for use in various ‘click’ reactions have been reported and have been used to form macro-RAFT agents. These RAFT agents include those with active ester or thiazolidine-2-thione functionality for reaction with amine functional substrates, with alkyne or azide functionality (for use in copper catalyzed cycloaddition), or with various thiol reactive groups.
Sumerlin and co-workers[406] prepared a macro-RAFT agent by conjugation of RAFT agent 131 (with active ester functionality) with free amino groups of lysozyme. This was used in RAFT polymerization to attach poly(NIPAm)-b-poly(DMAm) by sequential monomer addition. In another study they[420] prepared a macro-RAFT agent by conjugation of a RAFT agent having ene-functionality (140) to a free thiol group of bovine serum albumin (BSA). This was used in RAFT polymerization to attach a poly(NIPAm) block.
Biopolymer Conjugates Post-RAFT Polymerization
Interest in the synthesis of block and graft copolymers (conjugates) with biological polymers continues unabated. Methods for preparing biopolymer conjugates have been reviewed.[53,54,57,89] The most common method involves coupling the RAFT-synthesized polymer with the biopolymer through reactive functionality present on either the ‘R’ or ‘Z’ groups. Note that use of functionality on ‘Z’ leaves the thiocarbonylthio group as a potentially degradable link at the block juncture. This may or may not be advantageous depending on the application. Most of the methods for conjugation have already been mentioned under end group transformation. They include the active ester–amine reaction, the thiazolidine-2-thione–amine reaction, the pyridyl disulfide–thiol reaction, and the thiol-ene or thio Michael reaction.
a) The Active Ester–Amine Reaction (Scheme 17)

|
This usually involves the use of a biopolymer (e.g. a protein) with primary amine groups and a RAFT-synthesized polymer bearing an active ester substituent. RAFT agents with active ester functionality include 25[259] and 130 (used to prepare poly(NIPAm) for conjugation with lysozyme).[386] The active ester may also be formed post-RAFT polymerization from a RAFT-synthesized polymer with a carboxy end-group.[382] Many studies have shown that active ester groups such as those present in 25 and 130 are substantially more reactive than the RAFT agent thiocarbonyl functionality and conditions can be found such that the active ester–amine reaction occurs to the exclusion of any thiocarbonylthio aminolysis. Nonetheless, a potential issue with this chemistry is that primary amines may react with the RAFT agent functionality instead of or as well as the active ester functionality. Similar concerns exist in the case of the thiazolidine-2-thione-amine reaction below.
b) Thiazolidine-2-thione–Amine Reaction (Scheme 18)

|
RAFT agents with thiazolidine-2-thione functionality include 24,[238] 107,[372] 109,[376] and 195 (used to prepare poly(HPMAm) for conjugation with lysozyme)[372] and 108 (used to prepare poly(PEGMA) for conjugation with lysozyme).[374] Comments are similar to those for the active ester amine reaction.
c) Pyridyl Disulfide–Thiol Reaction (Scheme 19)
|
|
This usually involves reaction between a RAFT-synthesized polymer bearing a pyridyl disulfide functionality with a thiol-functional biopolymer (e.g. a cystein residue). RAFT agents with pyridyl disulfide functionality include 86 (used to prepare poly(PEGA) to conjugate a thiol-functional peptide),[334] 164 (used to prepare poly(PEGA) to conjugate siRNA[452] and poly(NIPAm) which was coupled to glutathione[453]) 163 and 165 (used in preparing a trehalose glycopolymer which was coupled with a thiolated lysozyme), and 194 (used to synthesize poly(HPMAm) and thereby form a mid chain conjugate with BSA).[477] This pyridyl disulfide functionality is compatible with RAFT polymerization and most monomers. The pyridyl disulfide group can also be introduced post-polymerization by transformation of the RAFT end-group (see Click Reactions).
d) Thiol-ene or Thio-Michael Chemistry (Scheme 20)

|
The strategy involves some initial RAFT end-group transformation. The reactivity of thiol and ene functionality in radical polymerization is such that it must be introduced post-RAFT polymerization. Polymers with a thiol end groups can be formed by any of the methods mentioned above and conjugated to an ene-functional biopolymer. However, the alternative process of coupling an ene functional polymer with a thiol-functional biopolymer is more attractive because the greater availability of the latter.
Examples of conjugate formation are:
-
the reaction of poly(NIPAm) with a thiol end-group with maleimide-functional DNA.[247]
-
the reaction of poly(Am) with a thiol end-group with maleimide-functional oligodeoxyribonucleotide.[244]
-
thio-Michael reaction of poly(NIPAm) having a thiol end-group with a bis-maleimide to provide a poly(NIPAm) bearing a maleimide end-group. This, in turn, was used in a second thio-Michael reaction with the thiol functionality of albumin (BSA) or ovalbumin (OVA).[398]
-
trapping the aminolysis product poly(PEGA) dithiobenzoate with divinyl sulfone and using this in a second thio-Michael reaction with the thiol functionality of albumin (BSA).[281]
-
the thiol end-group of poly(BzMA)-b-poly(HPMA) was reacted in situ with AA in a thio-Michael process to form a carboxy-terminated polymer which was conjugated to an amine-functional folate derivative by DCC coupling.[161]
e) Aminooxy-Ketone/Aldehyde Reaction (Scheme 21)

|
RAFT agents with BOC-protected aminooxy functionality include 85[334] and 131.[408] Vázquez-Dorbatt et al.[408] exploited the aminooxy-ketone/aldehyde reaction for preparing protein conjugates to poly(NIPAm) prepared with 131.
f) Azide-Alkyne Click Reaction (Scheme 22)

|
The process is mentioned under end-functional polymers. An example in the context of biopolymer conjugate formation is the coupling of alkyne end-functional poly(HEA), prepared by RAFT polymerization using trithiocarbonate (161), with an azide functional peptide.[448]
g) Attachment of Ligands that Show Specific Protein Binding (Scheme 23)

|
An example is that of biotin with streptavidin. RAFT agents containing a biotin moiety include 166 and 167.[454] This chemistry was also exploited in preparing a heterotelechelic conjugate from α-glutathione-ω-biotin-poly(NIPAm).[450]
Most of the approaches (a–g above) have also been used in making (non-biopolymer) block, star, and graft copolymers.
Multiblock Copolymers
Multiblock copolymers are linear polymers consisting of more than three covalently interconnected polymer segments based on two or more different polymers or copolymer segments. The synthesis of multiblock copolymers has been performed by three different strategies.
The more traditional approach involves the interconnection of preformed telechelic polymers with suitably reactive end-groups which can be ‘clicked’ together to form the multiblock (Table 21). In this context, several papers have appeared on the synthesis of biodegradable HPMAm multiblocks.[255,307,379]
A second strategy relies simply on sequential monomer addition. Hadjiantoniou et al.[622] synthesized a series of block copolymers from di- through to a pentablock comprising poly(DMAEMA) and poly(MMA) segments by sequential addition starting with poly(DMAEMA)-dithiobenzoate prepared with cumyl dithiobenzoate. A series of blocks (diblock through tetrablock) with azide end-groups were synthesized and used for modifying silica particles.[429]
The third strategy employs either multi-RAFT agents or cyclic RAFT (when multi-RAFT agents are formed in situ). New reports on multi-block synthesis using such RAFT agents are included in Table 22. Ebeling and Vana[604] prepared multiblock copolymers based on the multi-trithiocarbonate 441. Ebeling et al.[623] have also performed numerical simulations to predict the ideal molar mass distribution that should result from polymerizations using such agents.
Star Polymers
The literature on the synthesis of star polymers using the RAFT process continues to grow rapidly. In this section we consider syntheses that begin with a substrate containing multiple thiocarbonylthio groups of appropriate design, a multi-RAFT agent, and ‘grafting to’ syntheses that involve coupling of RAFT-made polymers to a core or arms of defined structure. Formation of star nano- or microgels by copolymerization with a divinyl monomer, by self-assembly and crosslinking of block copolymers or by growth from a crosslinked polymer or a nanoparticle core is covered in the next section, Microgels and Nanoparticles.
Multi-RAFT Agents
The multi-RAFT agent may in principle be a small organic compound, an organometallic complex, a dendrimer, a hyperbranched species, a macromolecular species, a particle or indeed any moiety possessing multiple thiocarbonylthio groups. Of these, macromolecular species and particles are considered in the section on Polymer Brushes below. Recent examples of star cores are shown in Table 4 (dithioesters), Table 8 (trithiocarbonates), and Table 10 (xanthates).
The first publications in this field[16] recognized two limiting forms of star (or graft/brush copolymer) growth depending on the orientation of the thiocarbonylthio group with respect to the core.
-
In the first strategy, the propagating radicals are linear chains that dissociate from the core. ‘Z’-connected RAFT agents (445, Scheme 24) are employed. The advantage of this strategy is that by-products from star–star coupling are unlikely. The thiocarbonylthio functionality is retained at the core of the star. A potential disadvantage of the ‘propagation away from core’ strategy is that reactions that cleave the thiocarbonylthio groups (e.g. aminolysis, thermolysis) cause destruction of the star structure (i.e. loss of the arms, Scheme 24). This feature can also be used to advantage in developing supported polymer syntheses or degradable network polymers (see Polymer Networks). A further potential issue is that the thiocarbonylthio functionality may become sterically inaccessible as polymerization proceeds.
-
In the second strategy most propagating radicals remain attached to the core and ‘R’-connected RAFT agents (446, Scheme 25) are used. Most thiocarbonylthio functionality remains on the periphery of the star. However, a linear macro-RAFT agent is released to the polymerization medium by the RAFT process. Since propagating radicals are attached to the core, termination by star–star coupling is a complication. Because the thiocarbonylthio groups are end-groups, they can be cleaved (e.g. by aminolysis) without destroying the star structure (Scheme 25).

|

|
The advantages and disadvantages of the two approaches are considered in detail in several papers and it is clear that the relative importance of the various factors mentioned above depends strongly on the particular monomer and RAFT agent used.
Luo et al.[625] have described the preparation of poly(NIPAm) ‘stars’ from a hyperbranched poly(glycidol) (HPG)-based macro-RAFT agent prepared as shown in Scheme 26.

Self-Condensing Vinyl Polymerization
A method for forming hyperbranched polymers by RAFT involves self-condensing vinyl polymerization. This involves copolymerization of monomers containing RAFT functionality. Examples of such RAFT inimers are 20,[253] 23,[460] 178,[460] and 230.[512] Star polymers can be formed by chain extension of the hyperbranched structure. The synthesis of St and acrylic polymers was achieved using dithiobenzoates, 20[253] or 23,[256] or trithiocarbonate, 178,[460] and VAc polymers using xanthate, 230.[512]
‘Grafting to’
Various combinations of RAFT, ATRP, click, ring opening polymerization, and other methods have been used to synthesize star polymers, including mikto-arm stars.
Microgels and Nanoparticles
In this section we consider formation of polymer microgels, stars, or nanoparticles either by self-assembly and crosslinking of RAFT-synthesized block copolymers (Table 23) or by RAFT-mediated radical crosslinking polymerization (Table 24).

|
The work of the McCormick group in the area of nanoparticle formation by block copolymer self assembly and crosslinking and in using the structures formed as drug delivery vehicles has been reviewed.[47,53] The temperature-responsive triblock copolymer, PEO-b-poly(APMAm)-b-poly(NIPAm), was synthesized by RAFT polymerization in aqueous medium.[380] At room temperature, the polymer is hydrophilic and exists as unimers in aqueous solution. Increasing the solution temperature above the lower critical solution temperature (LCST) of the poly(NIPAm) block leads to self-assembly into micelles with poly(NIPAm) in the core. The amine functionality of the poly(APMAm) shell was crosslinked with terephthaldicarboxaldehyde (TDA) at pH 9.0 to generate shell crosslinked micelles with cleavable imine linkages (Scheme 27). The material has potential application in pH triggered drug delivery.

|
One of the more popular methods for forming both microgels (nanogels) and core-crosslinked star polymers involves (co)polymerization of a divinyl-monomer using RAFT or another form of RDRP. Star polymers can be formed either through use of a linear macro-RAFT agent in initial microgel formation or by use of an initially formed microgel as a macro-RAFT agent in a subsequent RAFT polymerization. These approaches can be combined to form mikto-arm stars.
Examples of applying these strategies are included in Table 24. The conditions for the synthesis of well defined, low dispersity, ‘arm-first’ star copolymers based on poly(PEGA), poly(NIPAm), and poly(tBuA) have been explored. For systems which generate poorly soluble crosslinked cores, an efficient (close to quantitative) incorporation of arms into the star polymer structure and a low dispersity product was observed. In contrast, with a solvent compatible crosslinked core, the extent of incorporation of the arms was significantly reduced and molecular weight distributions were broader.
Lubrizol have undertaken the large scale commercialization of RAFT-mediated radical crosslinking polymerization in producing a star polymer rheology control agent for oil.[160]
Metal Nanoparticles
A wide variety of processes have been exploited with a view to preparing gold and other metal nanoparticles. A summary of the literature through to 2007 is contained in our recent review.[62] Some recent examples are as follows:
-
Azide functional gold nanoparticles were reacted with RAFT-synthesized alkyne-functional poly(4VP)[388] or poly(CMS)-b-poly(St).[447]
-
Core crosslinked micelles were formed from poly(MPC)-b-poly(DMAEMA/NIPAm) and HAuCl4·4H2O.[426]
-
Polymers (including poly(tBA) and poly(NIPAm)) prepared with trithiocarbonate 159 were used to coat gold nanoparticles.[445]
-
Diblock copolymer, poly(NIPAm)-b-poly(DMAEAm)[367] and PEGA[349] with trithiocarbonate ends were used to form colloidally stable gold nanoparticles.
-
Diblock copolymer poly(BA)-b-poly(NIPAm) with xanthate ends and a derived thiol were used to form colloidally stable gold nanoparticles.[112]
-
Thiol terminated methacrylamide polymers were prepared by post RAFT polymerization modification of poly(PFPMA).[226] These were used to form a library of gold nanoparticles.
-
Thiol terminated poly(St) grafted Fe3O4 nanoparticles were used in forming gold nanoparticles.[407]
-
End-groups of poly(St) prepared with 89,[338] poly(NIPAm) prepared with 251,[536] and poly(NVCL) prepared with 227[505] were functionalized with thio end-groups and these polymers were used in forming gold nanoparticles.
-
Single chain nanoparticles formed by photo-crosslinking of poly(DMAEMA/296) were used as nanoreactors in forming gold nanoparticles.[197]
Polymer Brushes/Graft Copolymers/Comb Polymers/Surface Modification
There have been several recent reviews on surface modification. Four approaches will be considered with reference to polymer surface modification.[627]
-
The ‘grafting from’ process making use of substrates where ‘R’ is bound to the surface (Scheme 28). It is possible to form such ‘R’ connected RAFT agents (448) directly and use these in ‘grafting from’. It is also possible to produce radicals on the surface (449) by irradiation, from attached initiator functionality, or by grafting through (Scheme 30) and thereby form the ‘R’ connected RAFT in situ during RAFT polymerization.
-
The ‘grafting from’ process making use of substrates where ‘Z’ is bound to the surface (Scheme 29).
-
The ‘grafting through’ which involves conducting a RAFT polymerization in the presence of a surface with monomer functionality (Scheme 30). The mechanism is then the same as shown in Scheme 28.
-
The ‘grafting to’ process in which a RAFT-synthesized polymer is attached to the surface.

|

|
|
|
Grafting-from Processes
Macro-RAFT agents used as substrates in grafting from process have been included in Table 25.

|
RAFT polymerization has also been used to prepare substrates for ‘grafting from’ by ATRP. The process involves copolymerization of monomer(s) containing ATRP initiator functionality (e.g. 416[587] and 417[203]). An example of this is shown in Scheme 31.[628]

Grafting-through Processes
This approach requires attaching monomer functionality to the substrate which may be:
-
A polymer chain (Fig. 19)
-
Particles and surfaces. Nanocomposite materials have been prepared by grafting through polymerization. Examples include:
-
Grafting through polymerization of glycomonomers to divinylbenzene based polymer microspheres in the presence of cumyl dithiobenzoate.[166]
-
Grafting through polymerization of MMA, St, and tBA with 2-((phenylcarbonothioyl)thio)acetic acid 61 as RAFT agent to gold or ITO surfaces functionalized with 454 by electrodeposition. The RAFT agent 61 is a poor RAFT agent and does not provide control over MMA polymerization, yet free poly(MMA) obtained under these conditions was reported to have a very narrow dispersity.[634]

|
The process of formation of polymers containing active ester functionality or pyridyl disulfide functionality as substrates for ‘grafting to processes’ (see above) might also be considered as examples of ‘grafting through’ processes.
Grafting-to Processes
One use of RAFT polymerization in the ‘grafting-to’ approach involves synthesis of polymers with reactive end-groups or block structures by RAFT polymerization which are then self-assembled and/or bonded to a particle, surface, or other structure.
A second way that RAFT is applied in a ‘grafting-to’ approach involves the use of RAFT copolymerization to synthesize a functional scaffold to which other polymers and or functionality are subsequently attached.
Covalent attachment to graphene has the drawback that the bonds formed may disrupt the conjugated structure thereby leading to compromised physical or electronic properties. Thus, ‘grafting to’ approaches that involve non-covalent attachment based on π–π stacking seem attractive. Pyrene end-functional poly(NIPAm),[455] poly(DMAEA), and PAA[456] were prepared using a pyrene end-functional RAFT agent which was used in forming graphene composites.
RAFT-synthesized polymers based on trithiocarbonate 149 with trimethoxysilane functionality on Z were grafted to silica particles.[432] Since only the chains with the RAFT end-group were grafted, the process provides a method of polymer purification. The grafted chains were shown to be living through chain extension experiments and could be cleaved from the support by aminolysis.
Hou et al.[594] have reported on grafting RAFT-synthesized polymers to fused silica particles functionalized with ‘Z’-connected RAFT agent by a radical exchange process. Degrafting was also accomplished by addition-fragmentation coupling. Further examples include:
-
The use of azide-alkyne click chemistry in coupling poly(VAc) (formed with 231) to azide-functional starch.[518]
-
The use of thiol-ene chemistry to attach a glycopolymer to divinylbenzene microspheres.[166]
-
RAFT-synthesized PAA (co)polymers as dispersants in preparing CdS quantum dot nanocomposites.[325]
-
The use of the thiocarbonyl hetero-Diels Alder reaction to couple poly(iBA) (formed with 57) to cyclopentadiene-functional cellulose.[295]
Polymer Networks
Polymer networks can be formed by crosslinking RAFT-synthesized homopolymers, block copolymers, or star polymers, or can be formed directly by a RAFT (co)polymerization in the presence of crosslinking monomer (e.g. a divinyl monomer such as EGDMA, MBAm, TEGDMA, DVB, or 418–425 (Fig. 16)). A wide variety of crosslinking processes have been explored. Polymer networks have been applied in controlled release applications (pharmaceuticals, agrochemicals) and as media for chromatography.[577,636]
Degradable Networks
A poly(AA/MMA) dithiobenzoate macro-RAFT agent was used in preparing an acid degradable pH responsive network with 455 (Chart 6) as crosslinking agent.[269]
Hydrogels
A PNVP xanthate macro-RAFT agent was used in preparing a PNIPAm hydrogel with MBAm as crosslinking agent.[491]
Porous Polymer Monoliths
These were prepared:
-
from MAA with EGDMA as crosslinker and dibenzyl trithiocarbonate (89) as RAFT agent.[636] The RAFT agent used is unlikely to provide good control over MAA polymerization. The monolith was formed in the presence of clenbuterol to provide molecular imprinting.
-
from EHMA with EGDMA as crosslinker and dibenzyl trithiocarbonate (89) as RAFT agent.[577] The RAFT agent is unlikely to provide good control over EHMA polymerization. The monolith was formed in the presence of propan-1-ol/butane-1,4-diol as porogen. The monolith was further modified by surface RAFT polymerization of glycerol monomethacrylate (289).
-
from RAFT-synthesized PLA-b-poly(St/386) crosslinked by metathesis polymerization.[413] Nanopores were formed by etching of PLA phase.
Molecular Imprinted Polymers
These were prepared (see also bullet one under monoliths above):
-
from MAA with EGDMA as crosslinker and either benzyl dithiobenzoate (54) or benzyl dithioisobutyrate (74) as RAFT agent.[291] Neither RAFT agent is likely to provide good control over MAA polymerization. Particles were formed in the presence of atrazine, to provide molecular imprinting, and acetonitrile as porogen.
-
from 4VP with EGDMA as crosslinker and cumyl dithiobenzoate (11) as RAFT agent.[177] Particles were formed in the presence of 2,4-dichlorophenoxyacetic acid, to provide molecular imprinting, and methanol/water as porogen.
Kinetics of Network Formation
A study of the kinetics of St/DVB copolymerization with S-thiobenzoyl thioglycolic acid (61) as RAFT agent and with BPO initiator at 80°C was carried out.[301] Delayed gelation, lower molecular weights pre-gelation, and higher degrees of swelling for the network prepared by RAFT polymerization were considered consistent with the molecular weight between crosslinks and the crosslink density being more homogeneous.
Methacrylic Networks
Addition of trithiocarbonate 79 was found to reduce the volumetric shrinkage stress in crosslinked multi(meth)acrylate networks without influence on the glass transition temperature.[322] The rate of polymerization was reduced compared with that for the similar polymerization in the absence of 79.
RAFT Extensible Networks
The reaction of a thiomalic acid based polyester with thiocarbonyl-bis-imidazole provided a network with trithiocarbonate crosslinks (456, Scheme 32).[603] The crosslinks were then extended by RAFT polymerization of MMA or St. The poly(MMA) or poly(St) was cleaved from the network by aminolysis and characterized by GPC. This indicated moderate control over MMA polymerization (Đ < 1.5) but poor control over St polymerization (Đ > 4). The latter result may have been compromized by oxidative coupling of the α,ω-thiolo-poly(St) product.

A gold nanoparticle network with trithiocarbonate crosslinks was formed from azide functional gold nanoparticles and the bis alkyne RAFT agent 82. The crosslinks were then extended by RAFT polymerization of NIPAm.[332]
US Patents on RAFT
Table 26 provides a list of granted US patents published during 2009-early 2012 and is an update to the list that appeared in the Encyclopedia of Polymer Science.[551] The list includes three divisionals of the original CSIRO RAFT patent.[637–639]

|
Conclusions
The last two years has seen a further substantial expansion in the number of papers on applications of RAFT polymerization. These applications cover a diverse range of areas ranging from biomedical and optoelectronic applications to coatings and rheology control agents. This has meant a reduction in the number of papers that explore RAFT polymerization as a technique but rather an increase as people seek both to improve the process and further define the intimate details of the RAFT mechanism. We anticipate these trends will continue.
Abbreviations
-
AcS 4-acetoxystyrene
-
AA acrylic acid
-
AAEMA 2-(acetoacetoxy)ethyl methacrylate
-
AEMA 2-aminoethyl methacrylate (hydrochloride)
-
AEMAm N-(2-aminoethyl)methacrylamide (hydrochloride)
-
AEP 2-(acryloyloxy)ethyl phosphate
-
AIBN azoisobutyronitrile
-
ACHN azobis(1-cyclohexanenitrile)
-
ACVA azobis(cyanovaleric acid)
-
Am acrylamide
-
AMA allyl methacrylate
-
AMPS 2-acrylamido-2-methylpropanesulfonic acid sodium salt (AMPS)
-
AN acrylonitrile
-
APMAm N-(3-aminopropyl)methacrylamide (hydrochloride)
-
ATRP atom transfer radical polymerization
-
BA butyl acrylate
-
BAm N-butylacrylamide
-
BD butadiene
-
BES 1-butyl ethenesulfonate
-
BMA butyl methacrylate
-
BzAm N-benzylacrylamide
-
BDSMA t-butyldimethylsilyl methacrylate
-
BP β-pinene
-
ClPEA 2-chloropropionyloxyethyl acrylate
-
BPO dibenzoyl peroxide
-
CHAm N-cyclohexylacrylamide
-
CCT catalytic chain transfer
-
CHMA cycylohexyl methacrylate
-
CMS 4-(chloromethyl)styrene
-
CMA cholesteryl methacrylate
-
CPA 3-chloropropyl acrylate
-
Đ molar mass dispersity = ratio of mass average to number average molar mass[721]
-
DA dodecyl acrylate
-
DAAm diacetone acrylamide (N-(1,1-dimethyl-3-oxobutyl)acrylamide)
-
DADMAC diallyldimethylammonium chloride
-
Dc 1-decene
-
DEAm N,N-diethylacrylamide
-
DEAEMA 2-(diethylamino)ethyl methacrylate
-
DEAPMAm N-(3-(diethylamino)propyl)methacrylamide (hydrochloride)
-
DEGDMA diethyleneglycol dimethacrylate
-
DEGMA (diethylene glycol monomethyl ether) methacrylate
-
DEHEA di(ethylene glycol) 2-ethylhexyl ether acrylate
-
DFHA decafluoroheptyl acrylate
-
DMAm N,N-dimethylacrylamide
-
DMAEA 2-(dimethylamino)ethyl acrylate
-
DMAEAm N-(2-(dimethylamino)ethyl)acrylamide
-
DMAEMA 2-(dimethylamino)ethyl methacrylate
-
DMAPMAm N-(3-(dimethylamino)propyl)methacrylamide (hydrochloride)
-
DPAEMA 2-(diisopropylamino)ethyl methacrylate
-
DVB divinylbenzene
-
EA ethyl acrylate
-
EAA ethyl α-acetoxyacrylate
-
E ethene
-
EEA ethoxyethyl acrylate
-
EGDMA ethyleneglycol dimethacrylate
-
EGMA (ethylene glycol monomethyl ether) methacrylate
-
EHA 2-ethylhexyl acrylate
-
EVC N-ethyl-3-vinylcarbazole
-
EVE ethyl vinyl ether
-
GMA glycidyl methacrylate
-
HA hexyl acrylate
-
HDDA hexane-1,6-diol diacrylate
-
HEA 2-hydroxyethyl acrylate
-
HEAm 2-hydroxyethylacylamide
-
HEMA 2-hydroxyethyl methacrylate
-
HMA hexylmethacrylate
-
HMS 4-(hydroxymethyl)styrene
-
HPMA 2-hydroxypropyl methacrylate
-
HPMAm N-2-hydroxypropyl methacrylamide
-
iBoA isobornyl acrylate
-
iBMA isobutyl methacrylate
-
Ip isoprene
-
IPhMA 4-iodophenyl methacrylate
-
iPOx 2-isopropenyl-2-oxazoline
-
IUPAC International Union of Pure and Applied Chemistry
-
LAM less activated monomers (includes vinyl monomers such as VAc, NVP and NVC)
-
Lim limonene
-
LMA dodecyl methacrylate
-
LPO didodecanoyl peroxide (dilauroyl peroxide)
-
MA methyl acrylate
-
MADIX macromolecular design by interchange of xanthate
-
MAEP 2-(methacryloyloxy)ethyl phosphate
-
MAH maleic anhydride
-
MAM more activated monomers (includes styrenes, acrylates, and acrylamides)
-
MAN methacrylonitrile
-
MBAm methylene-bis-acrylamide
-
MMA methyl methacrylate
-
MMBL γ-methyl-α-methylene-χ-butyrolactone
-
MPC methacryloyloxyethyl phosphorylcholine
-
MVK methyl vinyl ketone
-
NAPM N-acryloyl-l-proline methyl ester
-
NAPAM N-acryloyl-l-phenylalanine methyl ester
-
NAP N-acryloylpyrrolidine
-
NAS N-acryloyloxysuccinimide
-
NEMI N-ethylmaleimide
-
NES neopentyl ethenesulfonate
-
NIPAm N-isopropylacrylamide
-
NMMI N-methylmaleimide
-
NMP aminoxyl-(nitroxide-)mediated polymerization
-
NPMI N-phenylmaleimide
-
NMS N-methacryloyloxysuccinimide
-
NVC N-vinylcarbazole
-
NVCL N-vinylcaprolactam
-
NVP N-vinylpyrrolidone
-
NVPI N-vinylphthalimide
-
SMe 4-methylstyrene
-
OAm N-octylacrylamide
-
PAm N-propylacrylamide
-
PA propargyl acrylate
-
PAA α-propylacrylic acid (2-methylenepentanoic acid)
-
PCL poly(caprolactone)
-
PDMS poly(dimethylsiloxane)
-
PE polyethylene
-
PEG poly(ethylene glycol) monomethyl ether
-
PEGA (poly(ethylene glycol) monomethyl ether) acrylate
-
PEGMA (poly(ethylene glycol) monomethyl ether) methacrylate
-
PEO poly(ethylene oxide)
-
PFPMA pentafluorophenyl methacrylate
-
PFPVB pentafluorophenyl 4-vinylbenzoate
-
PFPVS pentafluorophenyl 4-vinylbenzenesulfonate
-
PFS pentafluorostyrene
-
PLA poly(lactic acid)
-
PMA propargyl methacrylate
-
PMVE perfluoro(methyl vinyl ether)
-
POSS isobutyl polyhedral oligomeric silsesquioxane
-
PVK phenyl vinyl ketone
-
PVS phenyl 4-vinylbenzenesulfonate
-
py pyridine
-
RAFT reversible addition–fragmentation chain transfer
-
RDRP reversible deactivation radical polymerization
-
ROMP ring-opening metathesis polymerization
-
ROP ring-opening polymerization
-
SB 4-(3-butenyl)styrene
-
siRNA short interfering ribonucleic acid
-
ssDNA single stranded DNA
-
SSO3Na sodium styrene-4-sulfonate
-
St styrene
-
StMe 4-methylstyrene
-
TBAm N-tert-butylacrylamide
-
tBA tert-butyl acrylate
-
TBDMS-OS 4-(tert-butyldimethylsilyloxy)styrene
-
tBS 4-(tert-butoxy)styrene
-
TEGDA triethylene glycol diacrylate
-
TEGMA (triethylene glycol monomethyl ether) methacrylate
-
TEGDMA triethylene glycol dimethacrylate
-
TESPMA 3-(triethoxysilyl)propyl methacrylate
-
TFEMA 2,2,2-trifluoroethyl methacrylate
-
TFPMA 2,2,3,3-tetrafluoropropyl methacrylate
-
TFPA 2,2,3,3-tetrafluoropropyl acrylate
-
THPA tetrahydropyran acrylate
-
TMSEA 2-(trimethylsilyloxy)ethyl acrylate
-
TMSEMA 2-(trimethylsilyloxy)ethyl methacrylate
-
TMAEMA 2-(trimethylamonium)ethyl methacrylate
-
TMAPMA 3-(trimethylamonium)propyl methacrylate
-
TPMMA triphenylmethyl methacrylate
-
VAc vinyl acetate
-
VB vinyl butyrate
-
VBA 4-vinylbenzaldehyde
-
VBSC 4-vinylbenzenesulfonyl chloride
-
VBDA (4-vinylbenzyl)dimethylamine
-
VBTAC (vinylbenzyl)trimethylammonium chloride
-
VBTPC (4-vinylbenzyl)trimethylphosphonium chloride
-
VBz vinyl benzoate
-
VF2 vinylidene fluoride
-
Vim 4-vinylimidazole
-
VPv vinyl pivalate
-
2VP 2-vinylypyridine
-
4VP 4-vinylpyridine
References
[1] G. Moad, D. H. Solomon, The Chemistry of Radical Polymerization 2006, 2nd edn (Elsevier: Oxford).[2] G. Moad, in Polymer Science: A Comprehensive Reference 2012, 2nd edn, Vol. 3, pp. 59–118 (Eds K. Matyjaszewski, M. Möller) (Elsevier BV: Amsterdam).
[3] V. Sciannamea, R. Jerome, C. Detrembleur, Chem. Rev. 2008, 108, 1104.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFehsLo%3D&md5=36bab6993bcf016b840112a2bada3526CAS |
[4] R. B. Grubbs, Polym. Rev. 2011, 51, 104.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltl2gsL8%3D&md5=f8baa4ad8c77b2859d1f3374fb00e23bCAS |
[5] L. Tebben, A. Studer, Angew. Chem. Int. Ed. 2011, 50, 5034.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtFymu78%3D&md5=024a5af11fd1931bfda0d3a8b6d0719fCAS |
[6] J. Nicolas, Y. Guillaneuf, D. Bertin, D. Gigmes, B. Charleux, in Polymer Science: A Comprehensive Reference 2012, pp. 277–350 (Eds K. Matyjaszewski, M. Möller) (Elsevier: Amsterdam).
[7] K. Matyjaszewski, Macromolecules 2012, 45, 4015.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XlsVaqs7w%3D&md5=d60f3a4b97cde753ed0ade79e3a028a1CAS |
[8] K. Matyjaszewski, J. Spanswick, in Polymer Science: A Comprehensive Reference 2012, pp. 377–428 (Eds K. Matyjaszewski, M. Möller) (Elsevier: Amsterdam).
[9] K. Satoh, M. Kamigaito, M. Sawamoto, in Polymer Science: A Comprehensive Reference 2012, pp. 429–461 (Eds K. Matyjaszewski, M. Möller) (Elsevier: Amsterdam).
[10] B. M. Rosen, V. Percec, Chem. Rev. 2009, 109, 5069.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1Cjs7%2FJ&md5=57639a86232c88009ea39c2bca19af0fCAS |
[11] T. Fukuda, A. Goto, in Polymer Science: A Comprehensive Reference 2012, pp. 119–157 (Eds K. Matyjaszewski, M. Möller) (Elsevier: Amsterdam).
[12] T. R. Darling, T. P. Davis, M. Fryd, A. A. Gridnev, D. M. Haddleton, S. D. Ittel, R. R. Matheson, G. Moad, E. Rizzardo, J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 1706.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXivFyks78%3D&md5=da5148b35534e735d3a61a181a33725eCAS |
[13] S. Penczek, G. Moad, Pure Appl. Chem. 2008, 80, 2163.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlWnsbjN&md5=4bf795b7b42029875bf335634afe4e20CAS |
[14] A. D. Jenkins, R. I. Jones, G. Moad, Pure Appl. Chem. 2010, 82, 483.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXivVShsbs%3D&md5=b3f1ba864ec4aedc5cbf9f4e096180bbCAS |
[15] J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 1998, 31, 5559.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXkvF2gs7k%3D&md5=7b317daf025894dcec376159155980ccCAS |
[16] T. P. Le, G. Moad, E. Rizzardo, S. H. Thang, WO9801478 1998.
[17] G. Moad, E. Rizzardo, S. H. Thang, Aust. J. Chem. 2005, 58, 379.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXltFymsL4%3D&md5=0464d702bb4d20d3f965209912e5cd9bCAS |
[18] G. Moad, E. Rizzardo, S. H. Thang, Aust. J. Chem. 2006, 59, 669.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFeqsr%2FM&md5=dbce39d0fb32872e6e9f9a55bde54681CAS |
[19] G. Moad, E. Rizzardo, S. H. Thang, Aust. J. Chem. 2009, 62, 1402.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVers7bN&md5=fb2b73394696eb457b81af596f1722b8CAS |
[20] G. Moad, E. Rizzardo, S. H. Thang, Mater. Matters 2010, 2.
| 1:CAS:528:DC%2BC3MXktFWjurg%3D&md5=ac92ec3c0006f2160ebe24e089ba25b3CAS |
[21] G. Moad, E. Rizzardo, S. H. Thang, Strem Chemiker 2011, 25, 2.
| 1:CAS:528:DC%2BC3MXkt1Kru74%3D&md5=843a9b2a4a8034dcff415e27f1cdfaacCAS |
[22] G. Moad, E. Rizzardo, S. H. Thang, in Polymer Science: A Comprehensive Reference 2012, 2nd edn, Vol. 3, pp. 181–226 (Eds K. Matyjaszewski, M. Möller) (Elsevier BV: Amsterdam).
[23] M. Destarac, Polym. Rev. 2011, 51, 163.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltl2gs7c%3D&md5=7ee04dd762e1b441f408f12c291823f6CAS |
[24] C. Barner-Kowollik, J. P. Blinco, M. Destarac, K. J. Thurecht, S. Perrier, in Encyclopedia of Radicals in Chemistry, Biology and Materials 2012, Vol. 4, pp. 1895–1929 (Eds C. Chatgilialoglu, A. Studer) (John Wiley & Sons: Chichester, UK).
[25] G. Moad, E. Bicciocchi, M. Chen, J. Chiefari, C. Guerrero-Sanchez, M. Haeussler, S. Houshyar, D. Keddie, E. Rizzardo, S. Thang, J. Tsanaktsidis, in “Progress in Controlled Radical Polymerization: Mechanisms and Techniques”, ACS Symposium Series Vol. 1100 2012, p. 243 (Eds K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky) (American Chemical Society: Washington, D.C.).
[26] B. Klumperman, E. T. A. van den Dungen, J. P. A. Heuts, M. J. Monteiro, Macromol. Rapid Commun. 2010, 31, 1846.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtleiu77K&md5=c778d6ebc5f90484779083d17d474e01CAS |
[27] T. Junkers, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 4154.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptFGgtbk%3D&md5=72a0447ad992798143303463a82715fcCAS |
[28] D. J. Keddie, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 2012, 45, 5321.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xnt1WqsL4%3D&md5=7ea365135119828d45ef10190dca01c7CAS |
[29] J. Barth, M. Buback, Macromol. React. Eng. 2010, 4, 288.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmt1eitrs%3D&md5=04077185f4bf6c94cd027c24a03db548CAS |
[30] W. L. A. Brooks, B. S. Sumerlin, Isr. J. Chem. 2012, 52, 256.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xls1ensLg%3D&md5=8f3d8ffb0e747f6c2331ad40c214fbcaCAS |
[31] W. L. A. Brooks, B. S. Sumerlin, in “Progress in Controlled Radical Polymerization: Mechanisms and Techniques”, ACS Symposium Series Vol. 1100 2012, p. 277 (Eds K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky) (American Chemical Society: Washington, D.C.).
[32] J. M. O’Donnell, Chem. Soc. Rev. 2012, 41, 3061.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XksVOhu70%3D&md5=1f02d1597c05337c658db3835558ad95CAS |
[33] H. Willcock, R. K. O’Reilly, Polym. Chem. 2010, 1, 149.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlt1OhtL0%3D&md5=216b8c507188c7b2666fb8583000250fCAS |
[34] G. Moad, E. Rizzardo, S. H. Thang, Polym. Int. 2011, 60, 9.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1Slt7rK&md5=28dd877fd4b6adc5e5acd0b406f84e41CAS |
[35] P. J. Roth, C. Boyer, A. B. Lowe, T. P. Davis, Macromol. Rapid Commun. 2011, 32, 1123.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptFKqsLo%3D&md5=6fd2df8c62b3ef2b669848b85009afdbCAS |
[36] M. A. Harvison, P. J. Roth, T. P. Davis, A. B. Lowe, Aust. J. Chem. 2011, 64, 992.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVegt7bF&md5=f88dc05bc2d67b96cb57ac26e3a9c4feCAS |
[37] B. Quiclet-Sire, S. Z. Zard, Pure Appl. Chem. 2011, 83, 519.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosFeqtrY%3D&md5=0191dc830daa99e6dd3ae5ef2087771cCAS |
[38] M. A. Harvison, A. B. Lowe, Macromol. Rapid Commun. 2011, 32, 779.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmslynurc%3D&md5=4b706d762495d951940c13275e5650f3CAS |
[39] M. H. Stenzel, Macromol. Rapid Commun. 2009, 30, 1603.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtF2hu7nP&md5=25aae231e8973481a8924f90a0106497CAS |
[40] C. Boyer, M. H. Stenzel, T. P. Davis, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 551.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1ejur%2FF&md5=6a1879c25f81b7ac95962a978401b585CAS |
[41] Z. An, Q. Qiu, G. Liu, Chem. Commun. 2011, 47, 12424.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVGrs7fF&md5=ebfe39d51b8f601c0f57cd35236eb311CAS |
[42] A. Gregory, M. H. Stenzel, Prog. Polym. Sci. 2012, 37, 38.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVaqt7%2FO&md5=4c359c1457a38d5e50cfe6ba30dd7a33CAS |
[43] Y. Kwak, R. Nicolay, K. Matyjaszewski, Aust. J. Chem. 2009, 62, 1384.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVers7bJ&md5=949dbf051ff2eac24815bb4f69790d4cCAS |
[44] R. Nicolaÿ, Y. Kwak, Isr. J. Chem. 2012, 52, 288.
| Crossref | GoogleScholarGoogle Scholar |
[45] A. E. Smith, X. Xu, C. L. McCormick, Prog. Polym. Sci. 2010, 35, 45.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotV2gtw%3D%3D&md5=54e7b2c55fff916f78127477baff2fb5CAS |
[46] S. E. York, A. W. York, C. L. McCormick, in “Polymeric Delivery of Therapeutics” ACS Symposium Series Vol. 1053 2010, p. 49 (Eds S. E. Morgan, R. Y. Lochhead) (American Chemical Society: Washington, D.C.).
[47] M. G. Kellum, A. E. Smith, X. Xu, C. L. McCormick, in “Controlled/Living Radical Polymerization: Progress in RAFT, DT, NMP & OMRP”, ACS Symposium Series Vol. 1024 2009, p. 195 (Ed. K. Matyjaszewski) (American Chemical Society: Washington, D.C.).
[48] M. Semsarilar, S. Perrier, Nat. Chem. 2010, 2, 811.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtF2ms7nJ&md5=f7d58c3d991e8b260f32edfe6042e5fcCAS |
[49] E. Zengeni, A. Samakande, P.C. Hartmann, in Polymer Nanocomposites by Emulsion and Suspension Polymerization 2011, pp 244–268 (Ed. V. Mittal) (Royal Society of Chemistry: Cambridge, UK).
[50] M. Beija, J.-D. Marty, M. Destarac, Prog. Polym. Sci. 2011, 36, 845.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltVKqs7w%3D&md5=7045fd39ad7ca8fd8a725c02d906b059CAS |
[51] R. Pfaendner, Polym. Degrad. Stab. 2010, 95, 369.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhvFyis7k%3D&md5=957b6b3c6cdf9bbbec48dd792bb42b26CAS |
[52] A. Gregory, M. H. Stenzel, Expert Opin. Drug Delivery 2011, 8, 237.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpsVymtg%3D%3D&md5=710969d10298af33ec6ceae7f48a1b8dCAS |
[53] D. Smith, A. C. Holley, C. L. McCormick, Polym. Chem. 2011, 2, 1428.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotFOktr8%3D&md5=2a37bdf2c679f2baeaa3d9d834b215edCAS |
[54] C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu, S. Perrier, Chem. Rev. 2009, 109, 5402.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFCqtbvP&md5=2f4b95b050a6ce8b464972d503a30d01CAS |
[55] H. G. Börner, in “Controlled/Living Radical Polymerization: Progress in RAFT, DT, NMP & OMRP”, ACS Symposium Series Vol. 1024 2009, p. 265 (Ed. K. Matyjaszewski) (American Chemical Society: Washington, D.C.).
[56] S. G. Boyes, M. D. Rowe, C.-C. Chang, D. H. Thamm, S. L. Kraft, J. F. Harmon, N. J. Serkova, A. P. Vogt, B. S. Sumerlin, in “Polymeric Delivery of Therapeutics”, ACS Symposium Series Vol. 1053 2010, p. 65 (Eds S. E. Morgan, R. Y. Lochhead) (American Chemical Society: Washington, D.C.).
[57] V. Bulmus, Polym. Chem. 2011, 2, 1463.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotFOktro%3D&md5=2056a8d82a021ffa06ea962a1c3e7b65CAS |
[58] N. A. A. Rossi, J. N. Kizhakkedathu, in “Polymeric Delivery of Therapeutics”, ACS Symposium Series Vol. 1053 2010, p. 103 (Eds S. E. Morgan, R. Y. Lochhead) (American Chemical Society: Washington, D.C.).
[59] D. S. H. Chu, J. G. Schellinger, J. Shi, A. J. Convertine, P. S. Stayton, S. H. Pun, Acc. Chem. Res. 2012, 45, 1089.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmsVSqug%3D%3D&md5=480e51474193a3e3f2878f05d48def36CAS |
[60] B. S. Sumerlin, ACS Macro Letters 2012, 1, 141.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFOhtrnE&md5=63b1b4e0101b510ab88959d1c5110c60CAS |
[61] M. Haeussler, G. Moad, E. Rizzardo, J. Chiefari, AustralAsian. J. Cosmet. Sci. 2012, 25, 22.
[62] G. Moad, M. Chen, M. Häussler, A. Postma, E. Rizzardo, S. H. Thang, Polym. Chem. 2011, 2, 492.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvFWgur4%3D&md5=91edbca9351b803166c4632695f321d8CAS |
[63] D. A. Shipp, Polym. Rev. 2011, 51, 99.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltl2gsL4%3D&md5=409aef5b6804532a4cfd74d00680a7a3CAS |
[64] M. L. Coote, in Rate Constant Calculation for Thermal Reactions 2012, pp. 283–304 (Eds H. DaCosta, M. Fan) (John Wiley & Sons: Hoboken, NJ).
[65] Y. Tu, Z. Cheng, Z. Zhang, J. Zhu, W. Zhang, N. Zhou, P. Ni, X. Zhu, Sci. China Chem. 2010, 53, 1605.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVGju7nM&md5=531e0a586e1f88282b76b59b2d042cecCAS |
[66] U. Mansfeld, C. Pietsch, R. Hoogenboom, C. R. Becer, U. S. Schubert, Polym. Chem. 2010, 1, 1560.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFChsL7P&md5=8d2eafeeec550fdb58db6d4bd84f56bdCAS |
[67] R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade, C. J. Hawker, Chem. Rev. 2009, 109, 5620.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlymtbvO&md5=3d720434cd9052c8ee6cd9871ac6c110CAS |
[68] A. J. Inglis, C. Barner-Kowollik, Macromol. Rapid Commun. 2010, 31, 1247.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptVejtLc%3D&md5=b82bc9723b3fc9dc72f8bf9e606b617dCAS |
[69] R. Fu, G.-D. Fu, Polym. Chem. 2011, 2, 465.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvFWgtbY%3D&md5=420956b4b4fb2834e27fc8130ad2bef2CAS |
[70] S. Slavin, J. Burns, D. M. Haddleton, C. R. Becer, Eur. Polym. J. 2011, 47, 435.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkt1ehu7s%3D&md5=6d938ddb9e5aba465ab164765cbef412CAS |
[71] A. B. Lowe, Polym. Chem. 2010, 1, 17.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlt1eltbo%3D&md5=95f6ca7bfd97dd22fba6f8cfa51ba76eCAS |
[72] N. Akeroyd, B. Klumperman, Eur. Polym. J. 2011, 47, 1207.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtFarsLY%3D&md5=70a0de883f0f71f5e8c66806d7b77e49CAS |
[73] M. A. Tasdelen, M. U. Kahveci, Y. Yagci, Prog. Polym. Sci. 2011, 36, 455.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlKltbc%3D&md5=18397357422cead59c6dac49c10cee8fCAS |
[74] K. Nakabayashi, H. Mori, Int. J. Polym. Sci. 2012, 2012,
| Crossref | GoogleScholarGoogle Scholar |
[75] S. Beghdadi, I. A. Miladi, D. Addis, H. B. Romdhane, J. Bernard, E. Drockenmuller, Polym. Chem. 2012, 3, 1680.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XotFOqtLY%3D&md5=5d25fd08281f637b3914c62201715db0CAS |
[76] M. D. Green, M. H. Allen, J. M. Dennis, D. S.-l. Cruz, R. Gao, K. I. Winey, T. E. Long, Eur. Polym. J. 2011, 47, 486.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkt1ehu7c%3D&md5=b1cfc6a27c7104cb83540abddac77e73CAS |
[77] B. Ameduri, Macromolecules 2010, 43, 10163.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVais7vK&md5=b888553eb27ff3dbf73bac3a452195c1CAS |
[78] S. R. S. Ting, G. Chen, M. H. Stenzel, Polym. Chem. 2010, 1, 1392.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtl2msLnM&md5=e467ef177eb8ae90d683f78e4d15028bCAS |
[79] Q. Yang, in Engineered Carbohydrate-Based Materials for Biomedical Applications 2011, pp. 119–141 (Ed. R. Narain) (John Wiley & Sons: Hoboken, NJ).
[80] C. R. Becer, Macromol. Rapid Commun. 2012, 33, 742.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xls1Wks78%3D&md5=53a07c7210840550302ba018a5f46f83CAS |
[81] C. G. Hardy, L. Ren, J. Zhang, C. Tang, Isr. J. Chem. 2012, 52, 230.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xitlygsrc%3D&md5=6a4d5f8bff61628eaffb7180ec1aeb5cCAS |
[82] M. C. Stefan, M. P. Bhatt, P. Sista, H. D. Magurudeniya, Polym. Chem. 2012, 3, 1693.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XotFOqtLk%3D&md5=82e829761d9b787d9c937db9dc8eadf6CAS |
[83] M. Beija, M.-T. Charreyre, J. M. G. Martinho, Prog. Polym. Sci. 2011, 36, 568.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlKlur4%3D&md5=26b6d0e83e2185f601790453bd5c2603CAS |
[84] O. J. Cayre, N. Chagneux, S. Biggs, Soft Matter 2011, 7, 2211.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivFSqu7c%3D&md5=710402d7ad92b0492015d746613c0bcfCAS |
[85] J.-F. Lutz, Adv. Mater. 2011, 23, 2237.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlvVCitr8%3D&md5=4de78a20c33d01561e3fdf9f5df63c1fCAS |
[86] H. Kakwere, S. Perrier, Polym. Chem. 2011, 2, 270.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXitVyiur0%3D&md5=41c908bde96506d30663b0b27f18ec74CAS |
[87] H. F. Gao, K. Matyjaszewski, Prog. Polym. Sci. 2009, 34, 317.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjsl2hsL8%3D&md5=22491f182a362131efe005ef9bc22de4CAS |
[88] Y. Zhao, L. Wang, A. Xiao, H. Yu, Prog. Polym. Sci. 2010, 35, 1195.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1eku7fJ&md5=48a331a660d9aa11fccbd97bbc60943dCAS |
[89] R. M. Broyer, G. N. Grover, H. D. Maynard, Chem. Commun. 2011, 47, 2212.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1arurg%3D&md5=5eff22d5b77d0f1ffede8ed8e6634947CAS |
[90] S. Dehn, R. Chapman, K. A. Jolliffe, S. Perrier, Polym. Rev. 2011, 51, 214.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltl2gs7k%3D&md5=7baf043433ef939fdd4c48d19420888bCAS |
[91] M. Barz, R. Luxenhofer, R. Zentel, M. J. Vicent, Polym. Chem. 2011, 2, 1900.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVOltbjF&md5=6171f31aa7908fd7f079b4f24c99ab41CAS |
[92] F. J. Xu, W. T. Yang, Prog. Polym. Sci. 2011, 36, 1099.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptVWgtLg%3D&md5=65462b751c080bd2d7f13bae7ed9b6b8CAS |
[93] G. N. Grover, H. D. Maynard, Curr. Opin. Chem. Biol. 2010, 14, 818.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFamurzM&md5=28f3031398a93be3fc8a6b32daab07b2CAS |
[94] M. Tizzotti, A. Charlot, E. Fleury, M. Stenzel, J. Bernard, Macromol. Rapid Commun. 2010, 31, 1751.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlaksLbE&md5=a342256ba6e23075eae8514f3b1a872cCAS |
[95] E. Malmström, A. Carlmark, Polym. Chem. 2012, 3, 1702.
| Crossref | GoogleScholarGoogle Scholar |
[96] B. Charleux, F. D’Agosto, G. Delaittre, in Hybrid Latex Particles 2011, Vol. 233, pp. 125–183 (Eds A.M. van Herk, K. Landfester) (Springer: Berlin).
[97] M. J. Monteiro, Macromolecules 2010, 43, 1159.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkt1ahtg%3D%3D&md5=264776ff58183655a9f145044748b127CAS |
[98] C. Ebner, T. Bodner, F. Stelzer, F. Wiesbrock, Macromol. Rapid Commun. 2011, 32, 254.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovFertA%3D%3D&md5=e575b3e9190c8747d17212928d385b8cCAS |
[99] K. Kempe, C. R. Becer, U. S. Schubert, Macromolecules 2011, 44, 5825.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovVOju7k%3D&md5=c731e3b36325471e671d6a2764db6d1dCAS |
[100] D. Grishin, I. Grishin, Russ. J. Appl. Chem. 2011, 84, 2021.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtFCmsb8%3D&md5=658157630503a31ea663d711949efd4cCAS |
[101] J. Spanswick, B. Pike, in “Controlled/Living Radical Polymerization: Progress in ATRP”, ACS Symposium Series Vol. 1023 2009, p. 385 (Ed. K. Matyjaszewski) (American Chemical Society: Washington, D.C.).
[102] R. Y. Lochhead, in “Polymeric Delivery of Therapeutics”, ACS Symposium Series Vol. 1053 2010, p. 3 (Eds S. E. Morgan, R. Y. Lochhead) (American Chemical Society: Washington, D.C.).
[103] G. Pound-Lana, B. Klumperman, in “Controlled/Living Radical Polymerization: Progress in RAFT, DT, NMP & OMRP”, ACS Symposium Series Vol. 1024 2009, p. 167 (Ed. K. Matyjaszewski) (American Chemical Society: Washington, D.C.).
[104] J. Chiefari, R. T. A. Mayadunne, C. L. Moad, G. Moad, E. Rizzardo, A. Postma, M. A. Skidmore, S. H. Thang, Macromolecules 2003, 36, 2273.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXitVemurs%3D&md5=177a2137dc017ac41edc8c1bbc67173aCAS |
[105] Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo, S. H. Thang, Macromolecules 2003, 36, 2256.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXitVemuro%3D&md5=be1b7c9c3c7f9a31c2251b589d41e6e5CAS |
[106] X. Gao, S. Zhu, J. Appl. Polym. Sci. 2011, 122, 497.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosFClu7c%3D&md5=62d752ebe6fa756b0541b5da33cc5bf0CAS |
[107] R. Ran, Z. Chen, X. L. Wang, J. Appl. Polym. Sci. 2009, 111, 2011.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhvFaltA%3D%3D&md5=a2046814cd5bffaf3e00fb29dc4f9310CAS |
[108] E. Bicciocchi, Y. K. Chong, L. Giorgini, G. Moad, E. Rizzardo, S. H. Thang, Macromol. Chem. Phys. 2010, 211, 529.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXislKqsrY%3D&md5=b95052bacb841e1acc54ddc9ac295d80CAS |
[109] S. Houshyar, D. Keddie, G. Moad, R. Mulder, S. Saubern, J. Tsanaktsidis, Polym. Chem. 2012, 3, 1879.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XotFOqt7Y%3D&md5=b15147e09b637db523e8fa6d5f405844CAS |
[110] O. I. Strube, L. Nothdurft, M. Drache, G. Schmidt-Naake, Macromol. Chem. Phys. 2011, 212, 574.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivVShsrk%3D&md5=207d6a3f1736f9ec50e965805bf5169bCAS |
[111] D. Rinaldi, T. Hamaide, C. Graillat, F. D’Agosto, R. Spitz, S. Georges, M. Mosquet, P. Maitrasse, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 3045.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlvV2js78%3D&md5=7a655a80df948bffb0e910d99c05e898CAS |
[112] S. p. Sistach, M. Beija, V. Rahal, A. Brûlet, J.-D. Marty, M. Destarac, C. Mingotaud, Chem. Mater. 2010, 22, 3712.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmsVCqsrY%3D&md5=0b8dfd266dd36eced780dc78b81dd906CAS |
[113] M. Destarac, A. Guinaudeau, R. Geagea, S. Mazieres, E. Van Gramberen, C. Boutin, S. Chadel, J. Wilson, J. Polym. Sci, Part A:. Polym. Chem. 2010, 48, 5163.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVKjs77K&md5=23ab860c12ef328751d9d246d1843080CAS |
[114] I. S. Altarawneh, M. Srour, V. G. Gomes, Polym.-Plast. Technol. Eng. 2007, 46, 1103.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlaqtrzJ&md5=947e7ebf7c6e108aaec68e72c3cbe0e5CAS |
[115] D. J. Keddie, C. Guerrero-Sanchez, G. Moad, R. Mulder, E. Rizzardo, S. H. Thang, Macromolecules 2012, 45, 4205.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmvFajtrs%3D&md5=6783d565c06efc9c4d07b4b979ca83d0CAS |
[116] I. Rodríguez-Sanchez, D. Glossman-Mitnik, E. Zaragoza-Contreras, J. Mol. Model. 2009, 15, 1133.
| Crossref | GoogleScholarGoogle Scholar |
[117] C. Y. Lin, M. L. Coote, Aust. J. Chem. 2009, 62, 1479.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVers7bL&md5=57ccd8cc1b789407a09fc4961df20170CAS |
[118] E. Chernikova, V. Golubev, A. Filippov, C. Y. Lin, M. L. Coote, Polym. Chem. 2010, 1, 1437.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtl2msLnK&md5=5eab60fb602eddc2fbe520a316e37d42CAS |
[119] I. Rodríguez-Sanchez, E. Zaragoza-Contreras, D. Glossman-Mitnik, J. Mol. Model. 2010, 16, 95.
| Crossref | GoogleScholarGoogle Scholar |
[120] D. Glossman-Mitnik, Res. J. Chem. Sci. 2011, 1, 6.
| 1:CAS:528:DC%2BC38XltFWgtA%3D%3D&md5=130bab0ad694a000399d3dbda388d227CAS |
[121] C. Y. Lin, M. L. Coote, Aust. J. Chem. 2011, 64, 747.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotFOqu70%3D&md5=706091fdb46c5c8cbe634b2ffa094737CAS |
[122] W. Meiser, J. Barth, M. Buback, H. Kattner, P. Vana, Macromolecules 2011, 44, 2474.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXktFenur8%3D&md5=5cf106cbc0ca9f38779068e098e80d00CAS |
[123] W. Meiser, M. Buback, Macromol. Rapid Commun. 2011, 32, 1490.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosFCls74%3D&md5=265f7d860bb175c2017f1995e86465eaCAS |
[124] J. Barth, M. Buback, W. Meiser, P. Vana, Macromolecules 2010, 43, 51.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtl2qtbrL&md5=309423f557f2df3eb6edcebd1dfc9a5fCAS |
[125] W. Meiser, M. Buback, J. Barth, P. Vana, Polymer 2010, 51, 5977.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVGit7nL&md5=addd23054b0bfa73c5be095c3693437aCAS |
[126] C. Barner-Kowollik, M. Buback, B. Charleux, M. L. Coote, M. Drache, T. Fukuda, A. Goto, B. Klumperman, A. B. Lowe, J. B. Mcleary, G. Moad, M. J. Monteiro, R. D. Sanderson, M. P. Tonge, P. Vana, J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 5809.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVylt7vI&md5=94db7c36ad9bcd2202019cc35a80a6e3CAS |
[127] I. Zapata-González, E. Saldívar-Guerra, J. Ortiz-Cisneros, Macromol. Theory Simul. 2011, 20, 370.
| Crossref | GoogleScholarGoogle Scholar |
[128] D. Konkolewicz, B. S. Hawkett, A. Gray-Weale, S. Perrier, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 3455.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXntVGitb0%3D&md5=38043f0247c4b275506fae7f4f815378CAS |
[129] S. R. S. Ting, T. P. Davis, P. B. Zetterlund, Macromolecules 2011, 44, 4187.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlvFSqu7s%3D&md5=bcb33b6fd993dfd1a6d2eeb03e69f67fCAS |
[130] W. Meiser, M. Buback, Macromol. Rapid. Commun. 2012, 33, 1273.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmtFCgsLs%3D&md5=6850ae5e9e9dfa6bc1656e99c4311903CAS |
[131] T. Junkers, C. Barner-Kowollik, M. L. Coote, Macromol. Rapid Commun. 2011, 32, 1891.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlels73K&md5=47a471116ad9163797d79a7572bec03aCAS |
[132] G. Moad, Y. K. Chong, R. Mulder, E. Rizzardo, S. H. Thang, in “Controlled/Living Radical Polymerization: Progress in RAFT, DT, NMP & OMRP”, ACS Symposium Series Vol. 1024 2009, p. 3 (Ed. K. Matyjaszewski) (American Chemical Society: Washington, D.C.).
[133] K. Suzuki, Y. Nishimura, Y. Kanematsu, Y. Masuda, S. Satoh, H. Tobita, Macromol. React. Eng. 2012, 6, 17.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlKgurfF&md5=2602700c6dc5211388c5473052030e28CAS |
[134] S. L. Brown, D. Konkolewicz, A. Gray-Weale, W. B. Motherwell, S. Perrier, Aust. J. Chem. 2009, 62, 1533.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVers7nJ&md5=974af17f9827dfdb79ad0e4957848bbfCAS |
[135] J. Krstina, C. L. Moad, G. Moad, E. Rizzardo, C. T. Berge, M. Fryd, Macromol. Symp. 1996, 111, 13.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXktFaqsA%3D%3D&md5=176190d7eeeb2d11a3df5609ef8f4549CAS |
[136] J. Krstina, G. Moad, E. Rizzardo, C. L. Winzor, C. T. Berge, M. Fryd, Macromolecules 1995, 28, 5381.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXmsFejt74%3D&md5=de37f71003577308e13a7950ff4ac560CAS |
[137] G. Moad, E. Rizzardo, S. H. Thang, Polymer 2008, 49, 1079.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitlels7c%3D&md5=03552fa69703a325d5ca3be6a3b68ce2CAS |
[138] J. P. A. Heuts, N. M. B. Smeets, Polym. Chem. 2011, 2, 2407.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtl2nsrbL&md5=253b4ce53234db2e446e8a3b60a9d381CAS |
[139] S. Slavin, K. McEwan, D. M. Haddleton, in Polymer Science: A Comprehensive Reference 2012, pp. 249–275 (Eds K. Matyjaszewski, M. Möller) (Elsevier: Amsterdam).
[140] C. Barner-Kowollik, T. Junkers, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 1293.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVeit74%3D&md5=1968802b9c23d238358ec5b983b81e9cCAS |
[141] L. Chen, L. Yan, Q. Li, C. Wang, S. Chen, Langmuir 2010, 26, 1724.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVKhsrfE&md5=313a1b7773cb8382a7305ca6332ee1cbCAS |
[142] S. Moessmer, M. Roch, B. Goebelt, S. Johann, Farbe Lack 2009, 115, 18.
| 1:CAS:528:DC%2BD1MXht1agsrfJ&md5=f461eb79d43cf5e56692a174fddbcdaeCAS |
[143] S. Moessmer, M. Roch, B. Goebelt, S. Johann, Eur. Coat. J. 2010, 28.
| 1:CAS:528:DC%2BC3cXivVaqurY%3D&md5=d5b218e598a448db0e53fc66830ef458CAS |
[144] R. Poli, in Polymer Science: A Comprehensive Reference 2012, pp. 351–375 (Eds K. Matyjaszewski, M. Möller) (Elsevier: Amsterdam).
[145] K. S. S. Kumar, Y. Gnanou, Y. Champouret, J.-C. Daran, R. Poli, Chem.–Eur. J. 2009, 15, 4874.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtVGrtbs%3D&md5=b39dbc1ebcbc7437c01561ca9b602cbfCAS |
[146] M. Hurtgen, A. Debuigne, C. Jérôme, C. Detrembleur, Macromolecules 2010, 43, 886.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtFKn&md5=196b4404c15a5897a597155f234a9466CAS |
[147] D. N. Bunck, G. P. Sorenson, M. K. Mahanthappa, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 242.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVyhsb3E&md5=9e87faded4dd12bf4596aba2aea0ba6cCAS |
[148] H. J. Jeon, J. H. Youk, Macromolecules 2010, 43, 2184.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhvFOrsbY%3D&md5=60f51c3472ad0a8c55034910b35cde7dCAS |
[149] S. Yamago, Chem. Rev. 2009, 109, 5051.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVGktbrE&md5=2789dc3c94aa17e5b9ed8a3cda17b258CAS |
[150] S. Yamago, Y. Nakamura, in Polymer Science: A Comprehensive Reference 2012, pp. 227–247 (Eds K. Matyjaszewski, M. Möller) (Elsevier: Amsterdam).
[151] M. Okubo, Y. Sugihara, Y. Kitayama, Y. Kagawa, H. Minami, Macromolecules 2009, 42, 1979.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXisVCjuro%3D&md5=66244686a6d9eb8982f521c69d84fe3fCAS |
[152] H. Moribe, Y. Kitayama, T. Suzuki, M. Okubo, Macromolecules 2011, 44, 263.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtlGq&md5=2c34df765935b4f95b6f729e9f78444bCAS |
[153] Y. Kitayama, A. Chaiyasat, M. Okubo, Macromol. Symp. 2010, 288, 25.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjsFSrsro%3D&md5=b5a7820f0be83c31655c743171f3e3b2CAS |
[154] J. Hasegawa, K. Kanamori, K. Nakanishi, T. Hanada, S. Yamago, Macromol. Rapid Commun. 2009, 30, 986.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnvV2msbc%3D&md5=617f3c67e1b7f850e5cb7eb1462b8491CAS |
[155] S.-I. Yusa, S. Awa, M. Ito, T. Kawase, T. Takada, K. Nakashima, D. Liu, S. Yamago, Y. Morishima, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 2761.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmsFalsr0%3D&md5=cc9e4a0cc0dc4874153442220d8d7199CAS |
[156] E. Kayahara, H. Yamada, S. Yamago, Chem.–Eur. J. 2011, 17, 5272.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkvF2iur8%3D&md5=97767b99bab247831a818c040b3428bcCAS |
[157] Q. Zhang, J. Li, Z. Liu, Y. Ni, Sci. China Chem. 2010, 53, 2318.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtl2lurzI&md5=ff1bb2278dd82fbe24ddc75c9ff03d1bCAS |
[158] S. Yamago, E. Kayahara, H. Yamada, React. Funct. Polym. 2009, 69, 416.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtlequ7s%3D&md5=34d163f8eb6d76b795840f79640e8ba5CAS |
[159] R. Yodice, J. R. Johnson, R. L. Beebe, A. J. Brzytwa, C. D. Hilker, WO2009035793A1 2009.
[160] A. J. Brzytwa, J. Johnson, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem) 2011, 52, 533.
| 1:CAS:528:DC%2BC3MXhtVOjt7zK&md5=5a4981550fbf3d224660e545f10288fcCAS |
[161] C. Wei, K. Wu, J. Li, W. Ma, J. Guo, J. Hu, C. Wang, Macromol. Chem. Phys. 2012, 213, 557.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XisFKhsr8%3D&md5=d03d5053751abdeb2613e789297cd503CAS |
[162] C. Li, J. He, Y. Zhou, Y. Gu, Y. Yang, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 1351.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFemu7k%3D&md5=01ef86cc21ac141f7a70de186c2431c3CAS |
[163] Y. Kim, M. H. Pourgholami, D. L. Morris, M. H. Stenzel, Macromol. Biosci. 2011, 11, 219.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1CntLw%3D&md5=989e32f4dda6fadbe3586f758088d4feCAS |
[164] P. Zhao, J. Jiang, B. Leng, H. Tian, Macromol. Rapid Commun. 2009, 30, 1715.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1yltr3J&md5=8b02364edc54298bd605ee9cc5b8d78bCAS |
[165] J. Jiang, B. Leng, X. Xiao, P. Zhao, H. Tian, Polymer 2009, 50, 5681.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVWnurvL&md5=8250fd30e66df0e437c8e5599ba6f183CAS |
[166] A. Pfaff, L. Barner, A. H. E. Müller, A. M. Granville, Eur. Polym. J. 2011, 47, 805.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkt1egsb4%3D&md5=6ea052068c0e8ea3ea429da13955952dCAS |
[167] A. B. J. Withey, G. Chen, T. L. U. Nguyen, M. H. Stenzel, Biomacromolecules 2009, 10, 3215.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtleqtLrF&md5=53473d6ec5a9aa2d4e127cd20dc5c53cCAS |
[168] A. M. Elsen, R. Nicola, K. Matyjaszewski, Macromolecules 2011, 44, 1752.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjtVKnsLs%3D&md5=0785e6497a2a0d6f6bff613bd1888030CAS |
[169] K. Nilles, P. Theato, Polym. Chem. 2011, 2, 376.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXitVyiu7Y%3D&md5=37b7a65c5845e4ac070c4727987d71d7CAS |
[170] Y. Tao, K. Satoh, M. Kamigaito, Macromol. Rapid Commun. 2011, 32, 226.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlsVyrsQ%3D%3D&md5=f4739802071fb98a234bf9c6bbd7d4d7CAS |
[171] Y. X. Bai, Y. B. Liu, Y. F. Li, Q. Zhang, Polym. Adv. Technol. 2012, 23, 581.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVWls7s%3D&md5=3ddb454d77b71013eb7c450ec30906dcCAS |
[172] C. Bressy, M. N. Nguyen, B. Tanguy, V. G. Ngo, A. Margaillan, Polym. Degrad. Stab. 2010, 95, 1260.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmslCnsL8%3D&md5=95a2a32f37fed385633e01c4e2bdffa7CAS |
[173] K. Ishitake, K. Satoh, M. Kamigaito, Y. Okamoto, Polym. Chem. 2012, 3, 1750.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XotFOqtbw%3D&md5=a53e797c8837d43ce0e6768d473c5b28CAS |
[174] H.-Y. Tian, J.-J. Yan, D. Wang, C. Gu, Y.-Z. You, X.-S. Chen, Macromol. Rapid Commun. 2011, 32, 660.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXksVCntrs%3D&md5=2b0b0ac85c8fd9443893a08fd78434fcCAS |
[175] B. Kaur, L. D’Souza, L. A. Slater, T. H. Mourey, S. Liang, R. H. Colby, W. T. Ford, Macromolecules 2011, 44, 3810.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlsVKrsr8%3D&md5=168151f4c72a450923df73e607cdfa2bCAS |
[176] C.-U. Lee, D. Roy, B. S. Sumerlin, M. D. Dadmun, Polymer 2010, 51, 1244.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjt1GjtLY%3D&md5=1ab9dcf9fd98a8f25f41e07545e38763CAS |
[177] G. Pan, B. Zu, X. Guo, Y. Zhang, C. Li, H. Zhang, Polymer 2009, 50, 2819.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXms1equ7Y%3D&md5=4a39de1a16444cf3f1bba15dbd07b2c9CAS |
[178] M. B. Milovanovic, M. Avramovic, L. Katsikas, I. G. Popovic, J. Serb. Chem. Soc. 2010, 75, 1711.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFGktLs%3D&md5=27f428e0c4f0ed19e66822fc226bd12aCAS |
[179] P. Gerstel, C. Barner-Kowollik, Macromol. Rapid Commun. 2011, 32, 444.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXisVaht7c%3D&md5=07b517df517ae05f3f400651f164917cCAS |
[180] S. Jana, A. Parthiban, C. L. L. Chai, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 1494.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFemu7g%3D&md5=4a10635fd049772b473d2095ccd7ee35CAS |
[181] M. Chen, G. Moad, E. Rizzardo, Aust. J. Chem. 2011, 64, 433.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkvVCrs7w%3D&md5=456830fbbf54ddeefe77b02863511ffeCAS |
[182] M. Dietrich, M. Glassner, T. Gruendling, C. Schmid, J. Falkenhagen, C. Barner-Kowollik, Polym. Chem. 2010, 1, 634.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlOhtb%2FO&md5=fd0e109e8717a18831cfb62cf5372d3fCAS |
[183] F. Yhaya, S. Binauld, M. Callari, M. H. Stenzel, Aust. J. Chem. 2012, 65, 1095.
| Crossref | GoogleScholarGoogle Scholar |
[184] I. Y. Perevyazko, A. Vollrath, C. Pietsch, S. Schubert, G. M. Pavlov, U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 2906.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xmt1Wns7o%3D&md5=0ae8bd6b38f68e34c260d2b3324d748aCAS |
[185] Y.-H. Ng, F. di Lena, C. L. L. Chai, Polym. Chem. 2011, 2, 589.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvFWgu70%3D&md5=75195d18672b0bbd1eb427d1118940d4CAS |
[186] A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan, S. P. Armes, J. Am. Chem. Soc. 2011, 133, 16581.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFCgsLfM&md5=d2b74decffe9f467b7f65b357a6d527bCAS |
[187] A. Blanazs, A. J. Ryan, S. P. Armes, Macromolecules 2012, 45, 5099.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XosFemtrc%3D&md5=b79cb947a5f4fe54ad82cff592a0937cCAS |
[188] M. L. T. de Sordi, E. de Oliveira da Silva, M. A. Ceschi, C. L. Petzhold, React. Funct. Polym. 2011, 71, 648.
| Crossref | GoogleScholarGoogle Scholar |
[189] Y. Zhu, Y. Zhou, Z. Chen, R. Lin, X. Wang, Polymer 2012, 53, 3566.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XptVyhurg%3D&md5=7b1a055961ee20203815233044f2e3f4CAS |
[190] M. M. Bloksma, C. Weber, I. Y. Perevyazko, A. Kuse, A. Baumgärtel, A. Vollrath, R. Hoogenboom, U. S. Schubert, Macromolecules 2011, 44, 4057.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlvFSrtrc%3D&md5=1f40c0d2bd73a218fb0a9e19c67443efCAS |
[191] C. Weber, T. Neuwirth, K. Kempe, B. Ozkahraman, E. Tamahkar, H. Mert, C. R. Becer, U. S. Schubert, Macromolecules 2012, 45, 20.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1SmtbjL&md5=dca3c3e7af98f70e0b3bcc3d70610ceaCAS |
[192] J. Moraes, T. Maschmeyer, S. Perrier, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 2771.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmsFalsLo%3D&md5=d25a725cc0e893873c526400a7335333CAS |
[193] M. A. Harvison, T. P. Davis, A. B. Lowe, Polym. Chem. 2011, 2, 1347.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnt1KgsLo%3D&md5=a3d04974c82fd2dbd7853233d6a7baf8CAS |
[194] M. Chen, G. Moad, E. Rizzardo, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 6704.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlGjurzE&md5=aea33b9b58bc39553c81ed74cc6ed7a6CAS |
[195] K. Bebis, M. W. Jones, D. M. Haddleton, M. I. Gibson, Polym. Chem. 2011, 2, 975.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvFWntr0%3D&md5=4ac2d23904755e4bf249e005260076f6CAS |
[196] Y. Liu, Y. Wang, D. Zhuang, J. Yang, J. Yang, J. Colloid Interface Sci. 2012, 377, 197.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmvVelu7c%3D&md5=49084d12371c9c376164523598139b95CAS |
[197] J. He, L. Tremblay, S. Lacelle, Y. Zhao, Soft Matter 2011, 7, 2380.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivFWjtrg%3D&md5=2923e7ad59224baf252148a3891a3c97CAS |
[198] A. M. Breul, J. Schäfer, C. Friebe, F. Schlütter, R. M. Paulus, G. Festag, M. D. Hager, A. Winter, B. Dietzek, J. Popp, U. S. Schubert, Macromol. Chem. Phys. 2012, 213, 808.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xjt1Oqur8%3D&md5=e2dc079c8c4e9b3a7026b04ef83471e9CAS |
[199] R. Nicolaÿ, Macromolecules 2012, 45, 821.
| Crossref | GoogleScholarGoogle Scholar |
[200] M. Riedel, J. Stadermann, H. Komber, F. Simon, B. Voit, Eur. Polym. J. 2011, 47, 675.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkt1egs7s%3D&md5=3b2c132da898b0b852baabb8ca5e874aCAS |
[201] R. Menzel, A. Breul, C. Pietsch, J. Schäfer, C. Friebe, E. Täuscher, D. Weiß, B. Dietzek, J. Popp, R. Beckert, U. S. Schubert, Macromol. Chem. Phys. 2011, 212, 840.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXksVehsb8%3D&md5=d1c32b85370e0c7629268b9e043dadb2CAS |
[202] W. Sriprom, C. Neto, S. Perrier, Soft Matter 2010, 6, 909.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXitlSgtrw%3D&md5=f7780dfa1d088b21b0a88f082d02797cCAS |
[203] J. Rzayev, Macromolecules 2009, 42, 2135.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXitFKjsLs%3D&md5=13d08fceab34d624e487cf2947b167cfCAS |
[204] Q. Luo, H. Zheng, Y. Peng, H. Gao, L. Lu, Y. Cai, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 6668.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlGjurzJ&md5=69835740e1b1f7e2e377848fc25e81c6CAS |
[205] Z. Zhang, W. Wang, Z. Cheng, J. Zhu, N. Zhou, Y. Yang, Y. Tu, X. Zhu, Macromolecules 2010, 43, 7979.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtV2ktbfN&md5=0fb7e4a79db9f524bac7c62f5fafcab7CAS |
[206] Z. Zhang, W. Wang, H. Xia, J. Zhu, W. Zhang, X. Zhu, Macromolecules 2009, 42, 7360.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVOrs7rL&md5=99cf51884bfbd897f71bb54a7af4da13CAS |
[207] A. Krieg, C. Pietsch, A. Baumgaertel, M. D. Hager, C. R. Becer, U. S. Schubert, Polym. Chem. 2010, 1, 1669.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFChsL%2FJ&md5=d2c6c4e3a5562851337f84598db03932CAS |
[208] M. A. Kostiainen, C. Pietsch, R. Hoogenboom, R. J. M. Nolte, J. J. L. M. Cornelissen, Adv. Funct. Mater. 2011, 21, 2012.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmslKlurs%3D&md5=0e2556e113ce02a19f8e2542f991fb35CAS |
[209] T. Janoschka, A. Teichler, A. Krieg, M. D. Hager, U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 1394.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtFSmt7o%3D&md5=eb3f0caf4acb25cabc485be722639429CAS |
[210] G. M. Pavlov, A. M. Breul, M. D. Hager, U. S. Schubert, Macromol. Chem. Phys. 2012, 213, 904.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XlslKltLw%3D&md5=02b0369822a119d400aac448230b9707CAS |
[211] C. Pietsch, R. Hoogenboom, U. S. Schubert, Angew. Chem. Int. Ed. 2009, 48, 5653.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXoslOqsr0%3D&md5=a01d44a10360a4c248a18536b1006832CAS |
[212] C. Ulbricht, C. R. Becer, A. Winter, U. S. Schubert, Macromol. Rapid Commun. 2010, 31, 827.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmsVOjurc%3D&md5=df50e1b3e464aa6f86530f81ddf0fb91CAS |
[213] C. D. Grande, M. C. Tria, G. Jiang, R. Ponnapati, Y. Park, F. Zuluaga, R. Advincula, React. Funct. Polym. 2011, 71, 938.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpvFWhurY%3D&md5=0f54e366f2d29e66999407ac97b4ee71CAS |
[214] A. C. C. Esteves, P. Hodge, T. Trindade, A. M. M. V. Barros-Timmons, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5367.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFCksLjI&md5=228e1b73f7bd3b0c55b3b869c71759d5CAS |
[215] J. M. Pelet, D. Putnam, Macromolecules 2009, 42, 1494.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhvVChu7k%3D&md5=523ec3152037f55286f4337421e2a990CAS |
[216] L. Liu, J. Zhang, W. Lv, Y. Luo, X. Wang, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 3350.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotlWnsLs%3D&md5=782df3b9990c31b4c88f0b5aa15fe8b2CAS |
[217] O. Samsonova, C. Pfeiffer, M. Hellmund, O. M. Merkel, T. Kissel, Polymers 2011, 3, 693.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltF2rurk%3D&md5=445fe7e56990d7631e3cc8697bc1525cCAS |
[218] A. H. Alidedeoglu, A. W. York, D. A. Rosado, C. L. McCormick, S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 3052.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXns1ajtr0%3D&md5=05981fea369e61f48c9b83e6e15015f6CAS |
[219] A. H. Alidedeoglu, A. W. York, C. L. McCormick, S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5405.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFCksLjE&md5=6f74911061d204b20a6c824a9eb39617CAS |
[220] C. Boyer, A. Granville, T. P. Davis, V. Bulmus, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 3773.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXntlGgsr0%3D&md5=8ba8f33bffb36a245f06258dc20eb898CAS |
[221] Z. Li, A. K. Serelis, W. F. Reed, A. M. Alb, Polymer 2010, 51, 4726.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1WlsLrP&md5=7287ac76d58e525a1f258d483bd259caCAS |
[222] N. Bhuchar, Z. Deng, K. Ishihara, R. Narain, Polym. Chem. 2011, 2, 632.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvFWnsr4%3D&md5=40baf569b84ba9c1a1f3198480238f82CAS |
[223] S. Venkataraman, W. L. Ong, Z. Y. Ong, S. C. Joachim Loo, P. L. Rachel Ee, Y. Y. Yang, Biomaterials 2011, 32, 2369.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovVejtw%3D%3D&md5=c574d5569b0c20d90241395f884d5ca4CAS |
[224] W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao, Z. An, Macromolecules 2011, 44, 2524.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvVartLg%3D&md5=231f47a29abcb829a93f401296dd3d05CAS |
[225] M. Allmeroth, D. Moderegger, B. Biesalski, K. Koynov, F. Rösch, O. Thews, R. Zentel, Biomacromolecules 2011, 12, 2841.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnslKisrc%3D&md5=130fc7a111842431f6c0095d3bc67837CAS |
[226] M. I. Gibson, M. Danial, H.-A. Klok, ACS Combinat. Sci. 2011, 13, 286.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivFyjsbc%3D&md5=1ab44ea4163e4ec66ce6cf148237e62dCAS |
[227] L. Nuhn, M. Hirsch, B. Krieg, K. Koynov, K. Fischer, M. Schmidt, M. Helm, R. Zentel, ACS Nano 2012, 6, 2198.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xjt1SqtLk%3D&md5=5b8c0baf0a973b4a9ddeae16212b9ad5CAS |
[228] G. B. H. Chua, P. J. Roth, H. T. T. Duong, T. P. Davis, A. B. Lowe, Macromolecules 2012, 45, 1362.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsFyltL4%3D&md5=ce9a3de7158a37118afd1d6f6cae66a4CAS |
[229] Y. Li, S. P. Armes, Angew. Chem. Int. Ed. 2010, 49, 4042.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmvFSlurk%3D&md5=0ef484fa87cfedbaaaa4a29b1c15524eCAS |
[230] Y. Luo, L. Liu, X. Wang, H. Shi, W. Lv, J. Li, Soft Matter 2012, 8, 1634.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XlvVGqsA%3D%3D&md5=7eeb2c172fca31bc6192b95f593e23f9CAS |
[231] S. G. Spain, N. R. Cameron, Polym. Chem. 2011, 2, 1552.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotFOkt7Y%3D&md5=0c1ab76ea1a5597aeaded2dc28eef18aCAS |
[232] H. Kitano, A. Hayashi, H. Takakura, H. Suzuki, N. Kanayama, Y. Saruwatari, Langmuir 2009, 25, 9361.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnt1WkurY%3D&md5=7500a9c374a977b14a1a0f0072f06e59CAS |
[233] Z. Jia, J. Liu, T. P. Davis, V. Bulmus, Polymer 2009, 50, 5928.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCqur%2FK&md5=fee3010d3d5aaf587038c8dbb864c5e0CAS |
[234] S. Boissé, J. Rieger, A. Di-Cicco, P.-A. Albouy, C. Bui, M.-H. Li, B. Charleux, Macromolecules 2009, 42, 8688.
| Crossref | GoogleScholarGoogle Scholar |
[235] L. Wong, M. Kavallaris, V. Bulmus, Polym. Chem. 2011, 2, 385.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXitVyiu7c%3D&md5=ff2a9863ec65c8d256b6c1d1611e7f34CAS |
[236] M. Ahmed, R. Narain, Biomaterials 2011, 32, 5279.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtleht7s%3D&md5=bf7c1ed4b0c81ec9c61e4e273c01558cCAS |
[237] S. Kirkland-York, Y. Zhang, A. E. Smith, A. W. York, F. Huang, C. L. McCormick, Biomacromolecules 2010, 11, 1052.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjsl2qu7s%3D&md5=b18acd95f52d31b142f63cf55f060a0eCAS |
[238] L. Tao, J. Liu, J. Xu, T. P. Davis, Org. Biomol. Chem. 2009, 7, 3481.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpsl2it74%3D&md5=cb9989102ffab7aef8c4747935c4e506CAS |
[239] C. Rodriguez-Emmenegger, B. V. K. J. Schmidt, Z. Sedlakova, V. Šubr, A. B. Alles, E. Brynda, C. Barner-Kowollik, Macromol. Rapid Commun. 2011, 32, 958.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntVGks7g%3D&md5=bab074fbe51f0adca70a3205a6f2798eCAS |
[240] N. A. Cortez-Lemus, R. Salgado-Rodríguez, A. Licea-Claveríe, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 3033.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXns1ajtrc%3D&md5=17878ccb69c4fc7b73c72ee7d19a9af5CAS |
[241] J. Xu, L. Tao, C. Boyer, A. B. Lowe, T. P. Davis, Macromolecules 2010, 43, 20.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFaju7zM&md5=b3b42055fd997b1aad39193f60edec06CAS |
[242] D. Zhuang, J. Nie, J. Yang, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 1999.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsFCmsbc%3D&md5=7ab0c7a423d663c33d2290b87bccb0cbCAS |
[243] J. D. Flores, J. Shin, C. E. Hoyle, C. L. McCormick, Polym. Chem. 2010, 1, 213.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlt1OgtbY%3D&md5=3567aa582efead833ff0cf14f5c3a75aCAS |
[244] N. Kanayama, H. Shibata, A. Kimura, D. Miyamoto, T. Takarada, M. Maeda, Biomacromolecules 2009, 10, 805.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXisVKrs74%3D&md5=c92f260ef09a99e9168f7dac894305a2CAS |
[245] M. G. Kellum, A. E. Smith, S. K. York, C. L. McCormick, Macromolecules 2010, 43, 7033.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpsV2msbY%3D&md5=e28ebef4005543d679d421751bbcfb3bCAS |
[246] M. G. Kellum, C. A. Harris, C. L. McCormick, S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 1104.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVeitrY%3D&md5=1dc14da085371166c53cd0e3862d769eCAS |
[247] K. Isoda, N. Kanayama, D. Miyamoto, T. Takarada, M. Maeda, React. Funct. Polym. 2011, 71, 367.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1ersrw%3D&md5=a8e0f06a406eb22b801049ebabda9a99CAS |
[248] E. Chernikova, P. Terpugova, A. Filippov, E. Garina, V. Golubev, A. Gostev, E. Sivtsov, Russ. J. Appl. Chem. 2009, 82, 1882.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCjtLzN&md5=826c1e9fc41cd1486dbbe7272003d9a9CAS |
[249] R. Chandrawati, B. Städler, A. Postma, L. A. Connal, S.-F. Chong, A. N. Zelikin, F. Caruso, Biomaterials 2009, 30, 5988.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVGqtLnP&md5=91344292e29d52ac2f5e34f124c004c2CAS |
[250] P. Chytil, T. Etrych, J. Kríz, V. Subr, K. Ulbrich, Eur. J. Pharm. Sci. 2010, 41, 473.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1Gjt7nL&md5=3ed4ae9dffc1ac48c2e8120b043a8681CAS |
[251] A. W. York, F. Huang, C. L. McCormick, Biomacromolecules 2010, 11, 505.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFKhuw%3D%3D&md5=d423e5139f9515575d5e2ae28533e480CAS |
[252] B. Bharatiya, S.-I. Yusa, V. Aswal, L. Abezgauz, D. Danino, P. Bahadur, Bull. Chem. Soc. Jpn 2011, 84, 1227.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xjt1Oktw%3D%3D&md5=9f5ef0d77c248a7816bd13ecc7235ce5CAS |
[253] Z. Wei, X. Hao, P. A. Kambouris, Z. Gan, T. C. Hughes, Polymer 2012, 53, 1429.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjtVOju78%3D&md5=a39db9eddff3fde74950d75a27143b34CAS |
[254] Z. Jia, J. Liu, C. Boyer, T. P. Davis, V. Bulmus, Biomacromolecules 2009, 10, 3253.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtF2qtL3P&md5=1219e889a9f2377e018897b6e4cfe0a5CAS |
[255] J. Yang, K. Luo, H. Pan, P. Kopecková, J. Kopecek, React. Funct. Polym. 2011, 71, 294.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1eku78%3D&md5=90f02584d9c802d64cc1f84545b2b0eaCAS |
[256] L. Tao, J. Liu, B. H. Tan, T. P. Davis, Macromolecules 2009, 42, 4960.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnslGqsbc%3D&md5=b57cd3ee216e7b2aa091ee8e30af0aa8CAS |
[257] J. Xu, L. Tao, C. Boyer, A. B. Lowe, T. P. Davis, Macromolecules 2011, 44, 299.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1SrsLvP&md5=806808c4a867e57d570c3bef19e551b6CAS |
[258] J. Xu, L. Tao, J. Liu, V. Bulmus, T. P. Davis, Macromolecules 2009, 42, 6893.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVWjsLfE&md5=5ac380c57a20e1521d5d92b4ddb67c91CAS |
[259] K. T. Wiss, P. Theato, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 4758.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVWgur%2FL&md5=d27ff221af3a43c087e316701fe7e464CAS |
[260] K. Zhang, L. Gao, Y. Chen, Polymer 2010, 51, 2809.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmslGntL8%3D&md5=fb00294f0c9435c5f484c26fc9403dd2CAS |
[261] A. H. Alidedeoglu, C. A. Harris, N. Martinez-Castro, A. W. York, C. L. McCormick, S. E. Morgan, in “Polymeric Delivery of Therapeutics”, ACS Symposium Series Vol. 1053 2010, p. 113 (Eds S. E. Morgan, R. Y. Lochhead) (American Chemical Society: Washington, D.C.).
[262] M. Müllner, A. Schallon, A. Walther, R. Freitag, A. H. E. Müller, Biomacromolecules 2010, 11, 390.
| Crossref | GoogleScholarGoogle Scholar |
[263] C. D. Grande, M. C. Tria, G. Jiang, R. Ponnapati, R. Advincula, Macromolecules 2011, 44, 966.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXoslyjsw%3D%3D&md5=22f61583f78d44364a6fb3a80028c390CAS |
[264] M. C. R. Tria, R. C. Advincula, Macromol. Rapid Commun. 2011, 32, 966.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnvFaktLs%3D&md5=f529a57aa43a90614f4cf26273277496CAS |
[265] B. Li, D. Majonis, P. Liu, M. A. Winnik, Can. J. Chem. 2011, 89, 317.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivVSntrc%3D&md5=d3d7077f65b47b8bb19efb22443a7e53CAS |
[266] Y. Zheng, W. Turner, M. Zong, D. J. Irvine, S. M. Howdle, K. J. Thurecht, Macromolecules 2011, 44, 1347.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhslGiu7Y%3D&md5=02d59fac2fbfa69fc6da100df7d0a826CAS |
[267] A. J. Inglis, P. Pierrat, T. Muller, S. Brase, C. Barner-Kowollik, Soft Matter 2010, 6, 82.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFKnsrfL&md5=a584bdfc70ff7d6a8232e2c1a706b8c9CAS |
[268] A. J. Inglis, M. H. Stenzel, C. Barner-Kowollik, Macromol. Rapid Commun. 2009, 30, 1792.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlGisLrI&md5=462561c51c38800756233d90316e5f40CAS |
[269] X. Sui, Z. Fu, Y. Shi, J. Appl. Polym. Sci. 2011, 121, 1860.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXks1WgtLY%3D&md5=88c056b08aef5b3093a7364f75c674c8CAS |
[270] W. Li, J. Li, J. Gao, B. Li, Y. Xia, Y. Meng, Y. Yu, H. Chen, J. Dai, H. Wang, Y. Guo, Biomaterials 2011, 32, 3832.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvVSrtL8%3D&md5=0f19bba5fe6924ad827e33a308d63e95CAS |
[271] J. Akimoto, M. Nakayama, K. Sakai, T. Okano, Biomacromolecules 2009, 10, 1331.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXkt1aqu7w%3D&md5=8d4799abbbbee25eac4f887f984aeea8CAS |
[272] J. Akimoto, M. Nakayama, K. Sakai, T. Okano, J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 7127.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVSnur%2FN&md5=0a1a67870e73afc45b68d486b3624bbcCAS |
[273] X. Zhang, M. Oddon, O. Giani, S. Monge, J.-J. Robin, Macromolecules 2010, 43, 2654.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXisFGmtbc%3D&md5=cd867e98607457cd0aa3db932d6e4275CAS |
[274] E. Chernikova, V. Yulusov, K. Mineeva, V. Golubev, E. Garina, Polym. Sci. Ser. B 2011, 53, 437.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVajsrjI&md5=4aee7516ec0bf40a23b84f842fc703a8CAS |
[275] J.-A. Raust, L. Houillot, M. Save, B. Charleux, C. Moire, C. l. Farcet, H. Pasch, Macromolecules 2010, 43, 8755.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht12jtbfI&md5=1389baabfdcfc035e58b85554efcf8cfCAS |
[276] J. M. J. Paulusse, R. J. Amir, R. A. Evans, C. J. Hawker, J. Am. Chem. Soc. 2009, 131, 9805.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnslegsrw%3D&md5=6eea2ed4cf18aa23ccdf142d994b5346CAS |
[277] T. Krivorotova, A. Vareikis, D. Gromadzki, M. Netopilík, R. Makuska, Eur. Polym. J. 2010, 46, 546.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXitlSit70%3D&md5=e25c915991c1c2c887231e138d8636b1CAS |
[278] S. Kim, B. Lee, S. Kim, J. Bang, J. Cho, Macromol. Chem. Phys. 2010, 211, 1188.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXntV2jt74%3D&md5=353b79226bab0110f9b1752d7c9431d1CAS |
[279] S. Chen, A. Bertrand, X. Chang, P. Alcouffe, C. Ladavière, J.-F. o. Gérard, F. r. Lortie, J. Bernard, Macromolecules 2010, 43, 5981.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXot1Ghsrs%3D&md5=6b7e49ad99a4c1e087a1a75ca9f9d9fbCAS |
[280] C. St Thomas, H. Maldonado-Textle, A. Rockenbauer, L. Korecz, N. Nagy, R. Guerrero-Santos, J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 2944.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmtVCitrc%3D&md5=7a6bb869a8075158336e81c1d8aed287CAS |
[281] G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto, H. D. Maynard, Macromolecules 2009, 42, 7657.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1Sqt7vO&md5=fd2cf17033156b80dd38716fc0c4a5daCAS |
[282] A. Verma, G. M. Murray, J. Funct. Biomater. 2012, 3, 1.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhslKhsL8%3D&md5=c50d33e5dd7996df14738e45d9e1fe95CAS |
[283] N. Zhou, Z. Zhang, J. Zhu, Z. Cheng, X. Zhu, Macromolecules 2009, 42, 3898.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlslOrtbg%3D&md5=e888135ea9df53d3c2720ba21484fbc3CAS |
[284] Y. Katsumoto, N. Kubosaki, Macromolecules 2008, 41, 5955.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXoslCrsbw%3D&md5=e8de46b8bc2d0856266eeeb726320e10CAS |
[285] A. Bertrand, S. Chen, G. g. Souharce, C. Ladavière, E. Fleury, J. Bernard, Macromolecules 2011, 44, 3694.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlsVKltLs%3D&md5=182172f218ea6f71b1c34ee4f703ee75CAS |
[286] C.-L. Chen, Y.-H. Lo, C.-Y. Lee, Y.-H. Fong, K.-C. Shih, C.-C. Huang, Inorg. Chem. Commun. 2010, 13, 603.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlsVGktLs%3D&md5=684a7590221da882f952f99636f22e61CAS |
[287] H. Mori, I. Tando, H. Tanaka, Macromolecules 2010, 43, 7011.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpslWhsbY%3D&md5=8d113f55574b4e68f01c2e8ccf9d93fdCAS |
[288] P. J. Roth, P. Theato, in “Non-Conventional Functional Block Copolymers”, ACS Symposium Series Vol. 1066 2011, p. 23 (Eds P. Theato, A. F. M. Kilbinger, E. B. Coughlin) (American Chemical Society: Washington, D.C.).
[289] H. Mori, S. Okabayashi, React. Funct. Polym. 2009, 69, 441.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtlequ7c%3D&md5=2106980333d6913d23c5d49ede302a70CAS |
[290] H. Mori, E. Kudo, Y. Saito, A. Onuma, M. Morishima, Macromolecules 2010, 43, 7021.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpvVGgsb0%3D&md5=fec28fcad2e684c0830e89a6b6b76139CAS |
[291] S. Xu, J. Li, L. Chen, Talanta 2011, 85, 282.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntFGqs7w%3D&md5=9d71acb7c44f3b7619501217ee089d04CAS |
[292] J. Skey, C. F. Hansell, R. K. O’Reilly, Macromolecules 2010, 43, 1309.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhvVGrtQ%3D%3D&md5=3605409380ed3566222445aa42f59795CAS |
[293] M. M. Stamenović, P. Espeel, W. V. Camp, F. E. Du Prez, Macromolecules 2011, 44, 5619.
| Crossref | GoogleScholarGoogle Scholar |
[294] A. J. Inglis, C. Barner-Kowollik, Polym. Chem. 2011, 2, 126.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmvVOlsA%3D%3D&md5=577511ab061811ea300b9b163414a62aCAS |
[295] A. S. Goldmann, T. Tischer, L. Barner, M. Bruns, C. Barner-Kowollik, Biomacromolecules 2011, 12, 1137.
| 1:CAS:528:DC%2BC3MXisFyitL8%3D&md5=843a4a6867056681ef5f81eb9805c7abCAS |
[296] E. Espinosa, M. Glassner, C. Boisson, C. Barner-Kowollik, F. D’Agosto, Macromol. Rapid Commun. 2011, 32, 1447.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXoslaqtLY%3D&md5=802594cda9d1bca67e0afb13d1e38fe7CAS |
[297] M. Glassner, G. Delaittre, M. Kaupp, J. P. Blinco, C. Barner-Kowollik, J. Am. Chem. Soc. 2012, 134, 7274.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xls1Sjtbs%3D&md5=6adf38ef3ebc14d3577e6d355410743fCAS |
[298] M. Glassner, J. P. Blinco, C. Barner-Kowollik, Polym. Chem. 2011, 2, 83.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmvVOntQ%3D%3D&md5=2833e3f53926aac4089c728247596e2fCAS |
[299] T. Masukawa, A. Yokoyama, T. Yokozawa, Macromol. Rapid Commun. 2009, 30, 1413.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVWiu7vO&md5=9c397772e408df703c004fb1f059898cCAS |
[300] C.-Y. Quan, D.-Q. Wu, C. Chang, G.-B. Zhang, S.-X. Cheng, X.-Z. Zhang, R.-X. Zhuo, J. Phys. Chem. C 2009, 113, 11262.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmvVeksLk%3D&md5=e8a81ba9cfe56b26926b2a18c0409f4fCAS |
[301] M. Roa-Luna, G. Jaramillo-Soto, P. V. Castañeda-Flores, E. Vivaldo-Lima, Chem. Eng. Technol. 2010, 33, 1893.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlansL3L&md5=1638ac53fe5dca231a12e6f1ec53dcddCAS |
[302] T. Antoun, A. Iraqi, L. Kergoat, L. Miozzo, A. Yassar, Macromol. Chem. Phys. 2011, 212, 1129.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmslOlt7c%3D&md5=58533d1eff57f8ccdcc313a34836d642CAS |
[303] A. Samakande, R. D. Sanderson, P. C. Hartmann, Eur. Polym. J. 2009, 45, 649.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhvFGku70%3D&md5=bf0c961bdb297d66818bdb5ce579cc7cCAS |
[304] K. S. Pafiti, E. Loizou, C. S. Patrickios, L. Porcar, Macromolecules 2010, 43, 5195.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmsVGitrg%3D&md5=86a2185f9f8b0fb68fb6cb93c306cc82CAS |
[305] K. Huang, D. P. Canterbury, J. Rzayev, Macromolecules 2010, 43, 6632.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptlymtbY%3D&md5=17626f893e77914436753f89e4ae3b27CAS |
[306] C. Zhu, S. Jung, S. Luo, F. Meng, X. Zhu, T. G. Park, Z. Zhong, Biomaterials 2010, 31, 2408.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpsV2rtA%3D%3D&md5=e84c295b9cde37d5a0e1662d024afb58CAS |
[307] H. Pan, J. Yang, P. Kopečková, J. i. Kopeček, Biomacromolecules 2011, 12, 247.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFGktbfI&md5=0dcf4f1e124ba992bc110faf1c7bfaaeCAS |
[308] Y. Li, Y. Zhang, D. Yang, J. Hu, G. Lu, X. Huang, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 2084.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkvVemsb0%3D&md5=c9ae2b2b07b1af4bc6141cb62e56d6abCAS |
[309] A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt, C. Barner-Kowollik, Macromolecules 2010, 43, 5515.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnt1Cms7o%3D&md5=1d5b07d415e355a18beaaf8b7bdec90eCAS |
[310] L. Yao, M. Z. Rong, M. Q. Zhang, Y. C. Yuan, J. Mater. Chem. 2011, 21, 9060.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnsVWhu7s%3D&md5=6680ff853501acf15e2e743c6aa26191CAS |
[311] C. Herfurth, D. Voll, J. Buller, J. Weiss, C. Barner-Kowollik, A. Laschewsky, J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 108.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1anu7rK&md5=d510b6c93741d67de949b1a4e5b4829bCAS |
[312] A. Kaiser, S. Brandau, M. Klimpel, C. Barner-Kowollik, Macromol. Rapid Commun. 2010, 31, 1616.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFKlsr7E&md5=e469e8b8298a6f4456da8ac66dfcdaa1CAS |
[313] Z. Yao, J.-S. Zhang, M.-L. Chen, B.-J. Li, Y.-Y. Lu, K. Cao, J. Appl. Polym. Sci. 2011, 121, 1740.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXks1Wgsr0%3D&md5=09ad22d6c101d9549b948ccbee1226b0CAS |
[314] Z. X. Wang, Q. H. Zhang, Y. T. Yu, X. L. Zhan, F. Q. Chen, J. H. Xiong, Chin. Chem. Lett. 2010, 21, 1497.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlSnu7fP&md5=39db398891d9c38dad15c7225ce3a7b6CAS |
[315] R. Wei, Y. Luo, Z. Li, Polymer 2010, 51, 3879.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpvFegsrc%3D&md5=5921bcdd47e76eb95c9811cf66c47160CAS |
[316] C. S. Biswas, V. K. Patel, N. K. Vishwakarma, V. K. Tiwari, B. Maiti, P. Maiti, M. Kamigaito, Y. Okamoto, B. Ray, Macromolecules 2011, 44, 5822.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnvFKhtb0%3D&md5=67925efc4bdccac86407fd52a8eb76deCAS |
[317] S. Rajaram, P. B. Armstrong, B. J. Kim, J. M. J. Frechet, Chem. Mater. 2009, 21, 1775.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXkslaksbc%3D&md5=a8f6e2b42f85e54db0487c00f94b895fCAS |
[318] J. Li, J. Ren, Y. Cao, W. Yuan, Polymer 2010, 51, 1301.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjt1Gjtbs%3D&md5=69e77953d570d9922162cd4ca3a7df9bCAS |
[319] C. Schmid, S. Weidner, J. Falkenhagen, C. Barner-Kowollik, Macromolecules 2012, 45, 87.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1GlsLzL&md5=aa218f378f84ff9327ba654982407b68CAS |
[320] E. V. Chernikova, P. S. Terpugova, A. A. Baskakov, A. V. Plutalova, E. S. Garina, E. V. Sivtsov, Polym. Sci. Ser. B 2010, 52, 119.
| Crossref | GoogleScholarGoogle Scholar |
[321] K. Chang, Z. T. Dicke, L. J. Taite, J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 976.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFalt7vK&md5=37df6241d3667d0c285e3d16eb7beb56CAS |
[322] D. Leung, C. N. Bowman, Macromol. Chem. Phys. 2012, 213, 198.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Oht7zE&md5=8e6a5b6c49b77ea53bdc00b08948d5c3CAS |
[323] T. Qu, A. Wang, J. Yuan, J. Shi, Q. Gao, Colloids Surf. B 2009, 72, 94.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXms1KrtLw%3D&md5=4660ccc83af272e0eeb0884abeecebcdCAS |
[324] J. Ma, C. Yang, J. Luo, T. Shen, M. Lu, J. Macromol. Sci. Part A 2012, 49, 321.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XksFCktbY%3D&md5=d28866de712282231766c5123cca066eCAS |
[325] P. Das, W. Zhong, J. Claverie, Colloid Polym. Sci. 2011, 1.
[326] A. Sogabe, J. D. Flores, C. L. McCormick, Macromolecules 2010, 43, 6599.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptVSltL4%3D&md5=76830fcd3f9947c3c24c6812a8d5b61eCAS |
[327] F. Ran, S. Nie, J. Li, B. Su, S. Sun, C. Zhao, Macromol. Biosci. 2012, 12, 116.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVelsQ%3D%3D&md5=cc0ac4ee607c35773c432ba87e998580CAS |
[328] A. F. Tominey, J. Liese, S. Wei, K. Kowski, T. Schrader, A. Kraft, Beilstein J. Org. Chem. 2010, 6, 66.
| Crossref | GoogleScholarGoogle Scholar |
[329] D.-X. Pang, W.-D. Liu, T. Li, L.-F. Fang, B.-K. Zhu, J. Appl. Polym. Sci. 2011, 119, 2953.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFajsLnN&md5=ba06f6b52071b108f124de377aa34672CAS |
[330] F. Ran, S. Nie, W. Zhao, J. Li, B. Su, S. Sun, C. Zhao, Acta Biomater. 2011, 7, 3370.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpslKltrw%3D&md5=4a14cdf96669b5c82250d9f9eaf3bc0fCAS |
[331] E. Chernikova, Z. Poteryaeva, S. Belyaev, E. Sivtsov, Russ. J. Appl. Chem. 2011, 84, 1031.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovVyms7g%3D&md5=2247b001349f0e8f4870d997ffffae0eCAS |
[332] X. Lian, J. Jin, J. Tian, H. Zhao, ACS Appl. Mater. Interfaces 2010, 2, 2261.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpsVKrsr4%3D&md5=b80517c195e1dc7f3e808ab0840e789eCAS |
[333] J. Jin, J. Tian, X. Lian, P. Sun, H. Zhao, Soft Matter 2011, 7, 11194.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVKhsb%2FP&md5=a9882e03d4ccb5377153298a6d2e535fCAS |
[334] G. N. Grover, J. Lee, N. M. Matsumoto, H. D. Maynard, Macromolecules 2012, 45, 4958.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XotlSnsb4%3D&md5=621423bd056293e96eb7d799cf6377aeCAS |
[335] A. R. Kiasat, R. Badri, S. Sayyahi, Chin. Chem. Lett. 2008, 19, 1301.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhvV2iuw%3D%3D&md5=19218426b2a88648886cc0478143301bCAS |
[336] G. Couture, B. Améduri, Eur. Polym. J. 2012, 48, 1348.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XntlSqu7Y%3D&md5=39dca232dce26c22c4d35c9c65020044CAS |
[337] A. Bivigou-Koumba, E. Görnitz, A. Laschewsky, P. Müller-Buschbaum, C. Papadakis, Colloid Polym. Sci. 2010, 288, 499.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXltlCnsg%3D%3D&md5=6be4f8b53a1147b7acb0732f4740caa6CAS |
[338] S.-k. Bae, S.-Y. Lee, S. C. Hong, React. Funct. Polym. 2011, 71, 187.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpsV2lug%3D%3D&md5=d5faf2d5c96c428485d3c61e3d26c5b9CAS |
[339] C. Lefay, D. Glé, M. Rollet, J. Mazzolini, D. Bertin, S. Viel, C. Schmid, C. Boisson, F. D’Agosto, D. Gigmes, C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 803.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1ejurzE&md5=28dd3f3286feae64321146565900894aCAS |
[340] D. Zehm, A. Laschewsky, P. Heunemann, M. Gradzielski, S. Prevost, H. Liang, J. P. Rabe, J.-F. Lutz, Polym. Chem. 2011, 2, 137.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmvVOltw%3D%3D&md5=7150b0db93154d42f25b1344d9d5df7cCAS |
[341] A. V. Serbin, E. N. Karaseva, I. V. Dunaeva, E. B. Krut’ko, Y. A. Talyzenkov, M. P. Filatova, E. V. Chernikova, Polym. Sci. Ser. B 2011, 53, 116.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltl2itr0%3D&md5=7bac0d23d2a4851b93f246d2cd72c89dCAS |
[342] A. Serbin, E. Karaseva, E. Chernikova, I. Dunaeva, E. Krutko, M. Filatova, A. Zezin, Macromol. Symp. 2010, 296, 80.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFSgtrbF&md5=20df254629833c933685fd197eb5e1d8CAS |
[343] B. V. K. J. Schmidt, M. Hetzer, H. Ritter, C. Barner-Kowollik, Macromolecules 2011, 44, 7220.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVylsrnP&md5=2697815a77a9e9c8761f83739c202d4cCAS |
[344] V. Shibaev, M. Ivanov, N. Boiko, E. Chernikova, Dokl. Chem. 2009, 427, 183.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVWqt7rP&md5=52bb2d0b89b3646c617208fc50f6178fCAS |
[345] K. Satoh, M. Matsuda, K. Nagai, M. Kamigaito, J. Am. Chem. Soc. 2010, 132, 10003.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXot1Wnurw%3D&md5=7fa71b2a41330e5f79d1d8f239f1268aCAS |
[346] N. Haridharan, R. Bhandary, K. Ponnusamy, R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 1491.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVCnsg%3D%3D&md5=a9e5c6064d461787247d22160a9ca8d0CAS |
[347] M. Häussler, P. Lok, M. Chen, J. Jasieniak, R. Adhikari, S. King, S. Haque, C. M. Forsyth, K. Winzenberg, S. E. Watkins, E. Rizzardo, G. J. Wilson, Macromolecules 2010, 43, 7101.
| Crossref | GoogleScholarGoogle Scholar |
[348] J. D. Flores, N. J. Treat, A. W. York, C. L. McCormick, Polym. Chem. 2011, 2, 1976.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVOltbnL&md5=1af3f18b5116c6c07f4f3fbdc814e18eCAS |
[349] J. Liu, E. Setijadi, Y. Liu, M. R. Whittaker, C. Boyer, T. P. Davis, Aust. J. Chem. 2010, 63, 1245.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpvVKksb8%3D&md5=7c2627a9eb2149bd4807675ce3e11f13CAS |
[350] C. E. Nelson, M. K. Gupta, E. J. Adolph, J. M. Shannon, S. A. Guelcher, C. L. Duvall, Biomaterials 2012, 33, 1154.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVylsrzE&md5=0f255eea28daf03ce67159e1b2ccd166CAS |
[351] A. E. Smith, X. Xu, S. E. Kirkland-York, D. A. Savin, C. L. McCormick, Macromolecules 2010, 43, 1210.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjs12gsA%3D%3D&md5=a479491d64113e872f27fd78b9aa5b90CAS |
[352] A. J. Convertine, C. Diab, M. Prieve, A. Paschal, A. S. Hoffman, P. H. Johnson, P. S. Stayton, Biomacromolecules 2010, 11, 2904.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1enurnI&md5=b305f887c7a8e19d9e899b9d1ac507f3CAS |
[353] G. Liu, Q. Qiu, W. Shen, Z. An, Macromolecules 2011, 44, 5237.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXns1KntLc%3D&md5=b6eeccdf8d1d4987876aa3c4dd6f1648CAS |
[354] X. Zhang, S. p. Boissé, W. Zhang, P. Beaunier, F. D’Agosto, J. Rieger, B. Charleux, Macromolecules 2011, 44, 4149.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlslKisrc%3D&md5=9edeac0721cd1c598c42672a765a1255CAS |
[355] Q. Qiu, G. Liu, Z. An, Chem. Commun. 2011, 47, 12685.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsV2ktLjJ&md5=fd2f53e2983bd07149bcaa9a029713beCAS |
[356] E. F. Crownover, A. J. Convertine, P. S. Stayton, Polym. Chem. 2011, 2, 1499.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotFOkt78%3D&md5=4c985913a9124533501599e12898be9aCAS |
[357] N. J. Treat, D. Smith, C. Teng, J. D. Flores, B. A. Abel, A. W. York, F. Huang, C. L. McCormick, ACS Macro Letters 2012, 1, 100.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFSjsbrN&md5=0670b5338c8a324f2fe1a27127776fb3CAS |
[358] L. C. Paslay, B. A. Abel, T. D. Brown, V. Koul, V. Choudhary, C. L. McCormick, S. E. Morgan, Biomacromolecules 2012,
| Crossref | GoogleScholarGoogle Scholar |
[359] A. E. Smith, X. Xu, D. A. Savin, C. L. McCormick, Polym. Chem. 2010, 1, 628.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlOhtb%2FM&md5=1caf1169dce0a8988fb6d5e41aed439cCAS |
[360] W. Zhang, F. D’Agosto, O. Boyron, J. Rieger, B. Charleux, Macromolecules 2011, 44, 7584.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFelu7vJ&md5=e3f54fdd1e111d8abdecd6e331e11b39CAS |
[361] X. Zhang, J. Rieger, B. Charleux, Polym. Chem. 2012, 3, 1502.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xms1aqtrs%3D&md5=1def208a24644893469d4a26eb39fac9CAS |
[362] X. Zhang, S. Boisse, C. Bui, P.-A. Albouy, A. Brulet, M.-H. Li, J. Rieger, B. Charleux, Soft Matter 2012, 8, 1130.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVahsA%3D%3D&md5=ffe535944c2ade8f219332e97b0009d1CAS |
[363] R. N. Johnson, R. S. Burke, A. J. Convertine, A. S. Hoffman, P. S. Stayton, S. H. Pun, Biomacromolecules 2010, 11, 3007.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1Cgs7vE&md5=9954328e79605679f344d52dc2094447CAS |
[364] L. Hosta-Rigau, R. Chandrawati, E. Saveriades, P. D. Odermatt, A. Postma, F. Ercole, K. Breheney, K. L. Wark, B. Städler, F. Caruso, Biomacromolecules 2010, 11, 3548.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVeru7zP&md5=3c0e003f8b4d21ffd63c45080ae13cb3CAS |
[365] S. Kumar, S. G. Roy, P. De, Polym. Chem. 2012, 3, 1239.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XkvV2nt7w%3D&md5=bc0f83acf3f5b1a9f1369962baa3dd87CAS |
[366] C. H. Hornung, C. Guerrero-Sanchez, M. Brasholz, S. Saubern, J. Chiefari, G. Moad, E. Rizzardo, S. H. Thang, Org. Proc. Res. Dev. 2011, 15, 593.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivFygs7w%3D&md5=15ca10d5c3401d3e38eac3e61c9cc890CAS |
[367] M. A. Nash, P. Yager, A. S. Hoffman, P. S. Stayton, Bioconjug. Chem. 2010, 21, 2197.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVSisLvP&md5=334924ee2a8c7177058fd0470036d0f2CAS |
[368] Z. Sun, Y. Luo, Soft Matter 2011, 7, 871.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVWhsL8%3D&md5=1ae5ccdd3549684d36db1e916d7ec510CAS |
[369] H. Chen, Y. Luo, Macromol. Chem. Phys. 2011, 212, 737.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjslCjt7s%3D&md5=a04df2f73b8ee0282155ff221e774b89CAS |
[370] R. Pötzsch, S. Fleischmann, C. Tock, H. Komber, B. I. Voit, Macromolecules 2011, 44, 3260.
| Crossref | GoogleScholarGoogle Scholar |
[371] E. Soto-Cantu, B. S. Lokitz, J. P. Hinestrosa, C. Deodhar, J. M. Messman, J. F. Ankner, S. M. Kilbey, Langmuir 2011, 27, 5986.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkvVOqtbo%3D&md5=1be073cc75de4d2aa2842a7646fa5f4aCAS |
[372] L. Tao, J. Xu, D. Gell, T. P. Davis, Macromolecules 2010, 43, 3721.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjslamsbw%3D&md5=960f189b565bd5fcc4ea1f87c302e8e8CAS |
[373] Y. Liu, M. Li, D. Wang, J. Yao, J. Shen, W. Liu, S. Feng, L. Tao, T. P. Davis, Aust. J. Chem. 2011, 64, 1602.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsF2rtLrJ&md5=5c60ebfb8e676f04ab66256cc28ef9e8CAS |
[374] L. Tao, J. Liu, J. Xu, T. P. Davis, Chem. Commun. 2009, 6560.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlalur3I&md5=31d5b1461900469bd89821a696d3b333CAS |
[375] W. Shen, Q. Qiu, Y. Wang, M. Miao, B. Li, T. Zhang, A. Cao, Z. An, Macromol. Rapid Commun. 2010, 31, 1444.
| Crossref | GoogleScholarGoogle Scholar |
[376] L. Tao, G. Chen, L. Zhao, J. Xu, E. Huang, A. Liu, C. P. Marquis, T. P. Davis, Chem. Asian J. 2011, 6, 1398.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmvVegurY%3D&md5=f1b438d7d2985a82be6bebb81f502481CAS |
[377] M. Luzon, C. Boyer, C. Peinado, T. Corrales, M. Whittaker, L. Tao, T. P. Davis, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 2783.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXms1yhsbk%3D&md5=684bf5a22fdc4e4bce47ab4bcf2e22b4CAS |
[378] M. Chen, G. Moad, E. Rizzardo, R.A. Evans, M. Haeussler, WO2009155657A1, 2009.
[379] K. Luo, J. Yang, P. Kopeckova, J. Kopecek, Macromolecules 2011, 44, 2481.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsVCrtbw%3D&md5=43cfcec3ecb9cd9b6acfcb1cca88e7ffCAS |
[380] X. Xu, J. D. Flores, C. L. McCormick, Macromolecules 2011, 44, 1327.
| 1:CAS:528:DC%2BC3MXisVWlsrs%3D&md5=c9a3018539a087c4772f41a58c04b512CAS |
[381] T. Boursier, I. Chaduc, J. Rieger, F. D’Agosto, M. Lansalot, B. Charleux, Polym. Chem. 2011, 2, 355.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXitVyiu7s%3D&md5=2d359d657f263093f0f4d4b32e28357eCAS |
[382] A. L. Golden, C. F. Battrell, S. Pennell, A. S. Hoffman, J. J. Lai, P. S. Stayton, Bioconjug. Chem. 2010, 21, 1820.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFKmtbzM&md5=18bf5f1d2b9508304b7a42cae416e90bCAS |
[383] J. D. Flores, X. Xu, N. J. Treat, C. L. McCormick, Macromolecules 2009, 42, 4941.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnsFWqtLk%3D&md5=31fca02eac0eca8bcf7a729e44c3319eCAS |
[384] N. Haridharan, K. Ponnusamy, R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 5329.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlCrsLvF&md5=e416d8ddfa9403c2113efc9a9f6754e7CAS |
[385] K. Skrabania, A. Laschewsky, H. v. Berlepsch, C. Böttcher, Langmuir 2009, 25, 7594.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjs1agsro%3D&md5=83991e7177b5d7195e955148171340bfCAS |
[386] H. Li, A. P. Bapat, M. Li, B. S. Sumerlin, Polym. Chem. 2011, 2, 323.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXitVyiu74%3D&md5=9280ffe8bfc19e942c149145bc7e601bCAS |
[387] N. Haridharan, R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 1021.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmsVektg%3D%3D&md5=f43fa1e0227e54c1504655c28ae59982CAS |
[388] T. Zhang, Y. Wu, X. Pan, Z. Zheng, X. Ding, Y. Peng, Eur. Polym. J. 2009, 45, 1625.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlvFOrs7Y%3D&md5=d26cfd9b886a964b02f657f1e0da7862CAS |
[389] K. Y. Mya, E. M. J. Lin, C. S. Gudipati, L. Shen, C. He, J. Phys. Chem. B 2010, 114, 9119.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnvVaqu7c%3D&md5=226a8e4f2f46e7ac57a133b4684e95cdCAS |
[390] J. He, B. Yan, L. Tremblay, Y. Zhao, Langmuir 2011, 27, 436.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFakur3J&md5=c88b3de545f8dd67bf875d50fa1927fcCAS |
[391] C. Diehl, P. Laurino, N. Azzouz, P. H. Seeberger, Macromolecules 2010, 43, 10311.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFaku7jL&md5=a696eca8305a18b83c224d4f502a2207CAS |
[392] D. Roy, A. Ullah, B. S. Sumerlin, Macromolecules 2009, 42, 7701.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1SnsbvM&md5=31bddc083171b846c91b1295072e6c75CAS |
[393] Y. Luo, X. Wang, B.-G. Li, S. Zhu, Macromolecules 2011, 44, 221.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtlag&md5=de3e44e3c2d19e6793203330eae8ee73CAS |
[394] W.-M. Wan, C.-Y. Pan, Polym. Chem. 2010, 1, 1475.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtl2msLbO&md5=bad8de06dc274afd55c73a1ff6265abbCAS |
[395] H. Kim, Y. J. Kang, S. Kang, K. T. Kim, J. Am. Chem. Soc. 2012, 134, 4030.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XisVGhsbg%3D&md5=079e761d0b2a1352b2c4f5426fea674dCAS |
[396] W. M. Gramlich, G. Theryo, M. A. Hillmyer, Polym. Chem. 2012, 3, 1510.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xms1aqtrk%3D&md5=99e7959bac8a53cedbf4c47a9e007ba3CAS |
[397] W.-D. He, X.-L. Sun, W.-M. Wan, C.-Y. Pan, Macromolecules 2011, 44, 3358.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXksFarsb8%3D&md5=e8ba48ccedb2662f6bbd39e0395911ffCAS |
[398] M. Li, P. De, H. Li, B. S. Sumerlin, Polym. Chem. 2010, 1, 854.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtl2ms7rE&md5=a5aa2b1a36453e524251cea0740c0f18CAS |
[399] J. W. Bartels, P. L. Billings, B. Ghosh, M. W. Urban, C. M. Greenlief, K. L. Wooley, Langmuir 2009, 25, 9535.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmsleisrs%3D&md5=a3ba37c11dd3f8aad504775db9392166CAS |
[400] S. Boissé, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li, B. Charleux, Chem. Commun. 2010, 46, 1950.
| Crossref | GoogleScholarGoogle Scholar |
[401] M. Grogna, R. Cloots, A. Luxen, C. Jerome, C. Passirani, N. Lautram, J.-F. Desreux, M. Collodoro, M.-C. De Pauw-Gillet, C. Detrembleur, Polym. Chem. 2011, 2, 2316.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1eis7rM&md5=b9802e8649e7f7ccd423a458f8469c75CAS |
[402] P. Espeel, F. Goethals, M. M. Stamenovic, L. Petton, F. E. Du Prez, Polym. Chem. 2012, 3, 1007.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjsFKgsrk%3D&md5=b09b6124bde934695de2f3afcbd5f219CAS |
[403] Q. Lou, M. A. Kishpaugh, D. A. Shipp, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 943.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXntlaitQ%3D%3D&md5=4d99e0404706c0d0624df1edebd72206CAS |
[404] L. Ouyang, L. Wang, F. J. Schork, Macromol. Chem. Phys. 2010, 211, 1977.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFait73K&md5=eb384a5f989992c877e0d29f243260bbCAS |
[405] P. De, S. R. Gondi, D. Roy, B. S. Sumerlin, Macromolecules 2009, 42, 5614.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmsFGlsrc%3D&md5=c1e495fea0c192ef794a01bbbdf53c30CAS |
[406] H. Li, M. Li, X. Yu, A. P. Bapat, B. S. Sumerlin, Polym. Chem. 2011, 2, 1531.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotFOkt7s%3D&md5=4bd590a5e2cf648888137b098f83322cCAS |
[407] J. Tian, F. Zheng, H. Zhao, J. Phys. Chem. C 2011, 115, 3304.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhslGhu74%3D&md5=c4fbedd1a2bd451ace187243dfce90aaCAS |
[408] V. Vázquez-Dorbatt, Z. P. Tolstyka, H. D. Maynard, Macromolecules 2009, 42, 7650.
| Crossref | GoogleScholarGoogle Scholar |
[409] T. Liu, S. Liu, Anal. Chem. 2011, 83, 2775.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXis1arur4%3D&md5=86c20f0a3a91ac024ce5e1b8fbdab50aCAS |
[410] C.-Q. Huang, C.-Y. Pan, Polymer 2010, 51, 5115.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlSgtrvO&md5=041c1cd8445b5024a741f6377f9d954fCAS |
[411] H.-N. Lee, Z. Bai, N. Newell, T. P. Lodge, Macromolecules 2010, 43, 9522.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlWhsrzN&md5=ff6e18751dd3ad58a78d18ea06d57a05CAS |
[412] E. J. W. Crossland, P. Cunha, S. Scroggins, S. Moratti, O. Yurchenko, U. Steiner, M. A. Hillmyer, S. Ludwigs, ACS Nano 2010, 4, 962.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVSitLk%3D&md5=703e640879fe90a70720f4b7650b5dcaCAS |
[413] L. Chen, M. A. Hillmyer, Macromolecules 2009, 42, 4237.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtVGltbY%3D&md5=01e34db3f9a8ee5ce4f03704eb1c853dCAS |
[414] M. Seo, M. A. Amendt, M. A. Hillmyer, Macromolecules 2011, 44, 9310.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVarsrzM&md5=5db652fae0dba4a7ab0865b5fc78de19CAS |
[415] A. D. Celiz, O. A. Scherman, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 5833.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVagu7zF&md5=92a27b190434c6f5a9c10c3d5a803abeCAS |
[416] M. J. Nasrullah, A. Vora, D. C. Webster, Macromol. Chem. Phys. 2011, 212, 539.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivVShsrY%3D&md5=cf3aa9fc4886e89bdbc9b4db8b7e9144CAS |
[417] Y. Guo, J. D. van Beek, B. Zhang, M. Colussi, P. Walde, A. Zhang, M. Kröger, A. Halperin, A. Dieter Schlüter, J. Am. Chem. Soc. 2009, 131, 11841.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXptlSmsLs%3D&md5=2c2931d3d5f68c6d2d7e6023a66defd3CAS |
[418] W. Zhang, A. H. E. Müller, Polymer 2010, 51, 2133.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXltFyrsro%3D&md5=8474b9820552e54920eb9a5b0443a913CAS |
[419] W. Lv, L. Liu, Y. Luo, X. Wang, Y. Liu, J. Colloid Interface Sci. 2011, 356, 16.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhvVWltL4%3D&md5=875b74d409a8b41fc241418c34271daaCAS |
[420] M. Li, H. Li, P. De, B. S. Sumerlin, Macromol. Rapid Commun. 2011, 32, 354.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhslygtLk%3D&md5=1fb64b36a290e4cb53247e5ddc6a245cCAS |
[421] Y. Li, E. Themistou, J. Zou, B. P. Das, M. Tsianou, C. Cheng, ACS Macro Letters 2012, 1, 52.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVKiurvI&md5=58be8507c0716b01cfe0d121777fec32CAS |
[422] J. Liu, G. Chen, M. Guo, M. Jiang, Macromolecules 2010, 43, 8086.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFKksr3E&md5=0aca9ac65167743be45b48d64f0696edCAS |
[423] J. Du, R. K. O’Reilly, Macromol. Chem. Phys. 2010, 211, 1530.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptVansb4%3D&md5=4a2fb66311f85d508db3f680e62b78b5CAS |
[424] N. Saetung, I. Campistron, S. Pascual, J.-C. Soutif, J.-F. Pilard, L. Fontaine, Eur. Polym. J. 2011, 47, 1151.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkvVKnt7g%3D&md5=fb67a773c7ce7e2154e0a72aea292ba2CAS |
[425] S.-I. Yusa, T. Endo, M. Ito, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 6827.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVymtbnI&md5=9106651c143f676c6ad730d73b228d2dCAS |
[426] F. Goto, K. Ishihara, Y. Iwasaki, K. Katayama, R. Enomoto, S.-i. Yusa, Polymer 2011, 52, 2810.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmvFSkuro%3D&md5=a55b56e4540f398e3c5db7409a325bdeCAS |
[427] S. R. S. Ting, E. H. Min, P. B. Zetterlund, M. H. Stenzel, Macromolecules 2010, 43, 5211.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmtVynsb0%3D&md5=eede900e2c9f19f2077685584bd864f0CAS |
[428] P. Escalé, S. R. S. Ting, A. Khoukh, L. Rubatat, M. Save, M. H. Stenzel, L. Billon, Macromolecules 2011, 44, 5911.
[429] Y. Huang, T. Hou, X. Cao, S. Perrier, Y. Zhao, Polym. Chem. 2010, 1, 1615.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFChsL7E&md5=785c4cf281bd4537b6dc5ce128768340CAS |
[430] G. Zhao, P. Zhang, C. Zhang, Y. Zhao, Polym. Chem. 2012, 3, 1803.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XotFOqtbs%3D&md5=44421f6222efc043863e752ca8611f3fCAS |
[431] M. Zong, K. J. Thurecht, S. M. Howdle, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 2009, 50, 211.
| 1:CAS:528:DC%2BC3cXislOrsL0%3D&md5=3d683083b24b516e62326652f4141273CAS |
[432] Y. Huang, Q. Liu, X. Zhou, S. Perrier, Y. Zhao, Macromolecules 2009, 42, 5509.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXos1CgsLg%3D&md5=cf2babc9098382bf0f16245cc858e19bCAS |
[433] M. Chen, M. Haeussler, G. Moad, E. Rizzardo, Org. Biomol. Chem. 2011, 9, 6111.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVWgsr7O&md5=380505bb5780220a0aa59e155b0843c7CAS |
[434] P. Cotanda, A. Lu, J. P. Patterson, N. Petzetakis, R. K. O’Reilly, Macromolecules 2012, 45, 2377.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjtVGit74%3D&md5=2cb3e672e359dfbfe446ca4a89a30554CAS |
[435] L. Zhang, K. Fang, S. Fu, X. Zhang, A. Tian, J. Appl. Polym. Sci. 2012, 125, 915.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVeltw%3D%3D&md5=e4211addd1603cf7ae73fc351e3a2e75CAS |
[436] A. J. Tilley, M. Chen, S. M. Danczak, K. P. Ghiggino, J. M. White, Polym. Chem. 2012, 3, 892.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjsFKgs70%3D&md5=0050dfa9ee077720665237b3b12506bbCAS |
[437] G. Bar-Nes, R. Hall, V. Sharma, M. Gaborieau, D. Lucas, P. Castignolles, R. G. Gilbert, Eur. Polym. J. 2009, 45, 3149.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1ylurvE&md5=d60e5e29a853c26f45c0fc670c2d000fCAS |
[438] Y. Chen, W. Luo, Y. Wang, C. Sun, M. Han, C. Zhang, J. Colloid Interface Sci. 2012, 369, 46.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtVGjtLk%3D&md5=73d5d29e87ae05810b3af437d17c83deCAS |
[439] A. M. Alb, M. F. Drenski, W. F. Reed, J. Appl. Polym. Sci. 2009, 113, 190.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltl2ru7w%3D&md5=cfdc954f1bf278f295be90fb496fe555CAS |
[440] J. P. Blinco, A. Greiner, C. Barner-Kowollik, S. Agarwal, Eur. Polym. J. 2011, 47, 111.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsV2gu7zM&md5=2ea538e760dc2be97b9ae84d55980982CAS |
[441] C. J. Dürr, S. G. J. Emmerling, A. Kaiser, S. Brandau, A. K. T. Habicht, M. Klimpel, C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 174.
| Crossref | GoogleScholarGoogle Scholar |
[442] D. Konkolewicz, C. K. Poon, A. Gray-Weale, S. Perrier, Chem. Commun. 2011, 47, 239.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFeqtbzN&md5=d151080b84031ebadb0c27e115e1319bCAS |
[443] N. M. Ahmad, B. Charleux, C. Farcet, C. J. Ferguson, S. G. Gaynor, B. S. Hawkett, F. Heatley, B. Klumperman, D. Konkolewicz, P. A. Lovell, K. Matyjaszewski, R. Venkatesh, Macromol. Rapid Commun. 2009, 30, 2002.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFektbvE&md5=a765c94c482cb41a9bddc1916f752a4dCAS |
[444] N. P. Truong, Z. Jia, M. Burges, N. A. J. McMillan, M. J. Monteiro, Biomacromolecules 2011, 12, 1876.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltFGmt7s%3D&md5=be072c38cafd6928d34270f3bf2bd636CAS |
[445] M. Liang, I. C. Lin, M. R. Whittaker, R. F. Minchin, M. J. Monteiro, I. Toth, ACS Nano 2010, 4, 403.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVynsbrK&md5=a605565c45d05834483b1f09bd87d50cCAS |
[446] T. H. Ho, M. Levere, J.-C. Soutif, V. Montembault, S. Pascual, L. Fontaine, Polym. Chem. 2011, 2, 1258.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnt1Kgsrc%3D&md5=d623c92e011bcec461ea6a3ee4a3b792CAS |
[447] J. Lim, H. Yang, K. Paek, C.-H. Cho, S. Kim, J. Bang, B. J. Kim, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 3464.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntV2qu70%3D&md5=ca16d39125e3c3b2f648557ef27c38a1CAS |
[448] H. Kakwere, R. J. Payne, K. A. Jolliffe, S. Perrier, Soft Matter 2011, 7, 3754.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkt1Gqurk%3D&md5=42f9be44c7c4db55d473aa50abba05d5CAS |
[449] C. J. Dürr, S. G. J. Emmerling, P. Lederhose, A. Kaiser, S. Brandau, M. Klimpel, C. Barner-Kowollik, Polym. Chem. 2012, 3, 1048.
| Crossref | GoogleScholarGoogle Scholar |
[450] X. Huang, C. Boyer, T. P. Davis, V. Bulmus, Polym. Chem. 2011, 2, 1505.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotFOkt7w%3D&md5=f976b5271c8459041fba1f2c28be5488CAS |
[451] R. J. Mancini, J. Lee, H. D. Maynard, J. Am. Chem. Soc. 2012, 134, 8474.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XlslOrsb0%3D&md5=8faeb215a364774f9b0c24a33e820d3aCAS |
[452] K. Gunasekaran, T. H. Nguyen, H. D. Maynard, T. P. Davis, V. Bulmus, Macromol. Rapid Commun. 2011, 32, 654.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXksVCnsbg%3D&md5=dceb5af0af7feda230416a88f7f152d5CAS |
[453] C.-W. Chang, T. H. Nguyen, H. D. Maynard, Macromol. Rapid Commun. 2010, 31, 1691.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlCktbfO&md5=fbeb2a511887791ff4ed243983f5a605CAS |
[454] K. L. Heredia, L. Tao, G. N. Grover, H. D. Maynard, Polym. Chem. 2010, 1, 168.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlt1Ohurs%3D&md5=d2940aeba251b39ca8c76a23e1da1d6cCAS |
[455] J. Liu, W. Yang, L. Tao, D. Li, C. Boyer, T. P. Davis, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 425.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFOqtbrN&md5=4a2a5199d3679799fccf042aa29d67fcCAS |
[456] J. Liu, L. Tao, W. Yang, D. Li, C. Boyer, R. Wuhrer, F. Braet, T. P. Davis, Langmuir 2010, 26, 10068.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlt1WrtLk%3D&md5=670a7b1cf56d8cada7b791dc2ec53a04CAS |
[457] C. R. Kinnane, G. K. Such, F. Caruso, Macromolecules 2011, 44, 1194.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFaltrg%3D&md5=b8253ac0d71ec11633e836d7b0e9d7d8CAS |
[458] S. Cheng, S. R. S. Ting, F. P. Lucien, P. B. Zetterlund, Macromolecules 2012, 45, 1803.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVekur8%3D&md5=da1d5450fc148e653525072d253ba346CAS |
[459] M. P. Robin, M. W. Jones, D. M. Haddleton, R. K. O’Reilly, ACS Macro Letters 2012, 1, 222.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1Oju77J&md5=8a1aab2af51efd410c40ac78be2a1048CAS |
[460] C. Zhang, Y. Zhou, Q. Liu, S. Li, S. b. Perrier, Y. Zhao, Macromolecules 2011, 44, 2034.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjtVKisr8%3D&md5=a232c73da41807670e8c0fe808293348CAS |
[461] J. Ferreira, J. Syrett, M. Whittaker, D. Haddleton, T. P. Davis, C. Boyer, Polym. Chem. 2011, 2, 1671.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpslKltL0%3D&md5=936bb49db339e26dc095730de12a8488CAS |
[462] J. Liu, H. Duong, M. R. Whittaker, T. P. Davis, C. Boyer, Macromol. Rapid Commun. 2012, 33, 760.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XlsFSqsLY%3D&md5=e63c347f3e0fe7fefcf1563c992a2034CAS |
[463] C. Boyer, M. Whittaker, T. P. Davis, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 5245.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Gqs7fI&md5=fffac885b44b6c78224a5f364608c33fCAS |
[464] E. H. Min, S. R. S. Ting, L. Billon, M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 3440.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotlWntr4%3D&md5=bfddc5a715ed0f9930430400b3a9d460CAS |
[465] S. Wallyn, M. Lammens, R. K. O’Reilly, F. D. Prez, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 2878.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmsFals7Y%3D&md5=bbb1397b9093192690a2c6484ee3a7f5CAS |
[466] S. Bhattacharjee, D. Bong, J. Polym. Environ. 2011, 19, 203.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltVKnsrs%3D&md5=a04cac30e0051f3f3a86b3064485aeeaCAS |
[467] K. Palaniappan, N. Hundt, P. Sista, H. Nguyen, J. Hao, M. P. Bhatt, Y.-Y. Han, E. A. Schmiedel, E. E. Sheina, M. C. Biewer, M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 1802.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjt1Giu78%3D&md5=0f3ac520198b11e4d5d5ce43edf191efCAS |
[468] C. Yang, J. K. Lee, A. J. Heeger, F. Wudl, J. Mater. Chem. 2009, 19, 5416.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXovVOisLw%3D&md5=fc71ace7f4dd63fbd6878aa624fdb6bdCAS |
[469] P. E. Williams, A. O. Moughton, J. P. Patterson, S. Khodabakhsh, R. K. O’Reilly, Polym. Chem. 2011, 2, 720.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvFWns70%3D&md5=8aa01cc3559cf00e1ade9d9780555f46CAS |
[470] A. C. Evans, J. Skey, M. Wright, W. Qu, C. Ondeck, D. A. Longbottom, R. K. O’Reilly, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 6814.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVymtbnN&md5=be3aeb95a6078ed1e8bf79374914a60eCAS |
[471] A. C. Evans, A. Lu, C. Ondeck, D. A. Longbottom, R. K. O’Reilly, Macromolecules 2010, 43, 6374.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXosFyqurg%3D&md5=efe74c888ed68156f767ebe32000857fCAS |
[472] A. O. Moughton, R. K. O’Reilly, Chem. Commun. 2010, 46, 1091.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlymsLc%3D&md5=811f2bfb01322d796103c69b2814e138CAS |
[473] A. O. Moughton, J. P. Patterson, R. K. O’Reilly, Chem. Commun. 2011, 47, 355.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFeqtLvI&md5=035ef323cac4ec40bcf97700db292799CAS |
[474] R. O’Reilly, C. Hansell, Polymers 2009, 1, 3.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptlSmtLo%3D&md5=4c3515ac347a624917a6ce8d2595db3bCAS |
[475] K. Skrabania, W. Li, A. Laschewsky, Macromol. Chem. Phys. 2008, 209, 1389.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXovFarsb4%3D&md5=4f35852381749d4f23ad1f001d57731eCAS |
[476] R. Rotzoll, P. Vana, Aust. J. Chem. 2009, 62, 1473.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVers7bP&md5=9c82e94e513394d8fb3abcc6d487fef3CAS |
[477] L. Tao, J. Liu, T. P. Davis, Biomacromolecules 2009, 10, 2847.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtV2ns7fI&md5=63803cacfe0f798d40a0dbed9f994548CAS |
[478] P. Fei, K. A. Cavicchi, ACS Appl. Mater. Interfaces 2010, 2, 2797.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtF2qsrvM&md5=6a408f06afca546a235ca9902da11472CAS |
[479] Y. He, T. P. Lodge, Chem. Commun. 2007, 2732.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmvV2mtbs%3D&md5=11cc82f6f9733e00bab6b9d7da391f67CAS |
[480] N. Saetung, I. n. Campistron, S. Pascual, J.-F. o. Pilard, L. Fontaine, Macromolecules 2011, 44, 784.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXoslOktw%3D%3D&md5=f34e08bd24e5c26e40c6dffce2e6c2a9CAS |
[481] L. Munuera, R. K. O’Reilly, Dalton Trans. 2010, 39, 388.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFKnsLjP&md5=63935fa6fcd3023ccc23d905d8a2f5a7CAS |
[482] M. Schwabe, R. Rotzoll, S. Küchemann, K. Nadimpalli, P. Vana, K. Samwer, Macromol. Chem. Phys. 2010, 211, 1673.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXps1Ghs7g%3D&md5=a5487f808510fcf47856245f78671b21CAS |
[483] L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo, H. D. Maynard, Macromolecules 2009, 42, 8028.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXptF2lsro%3D&md5=c3bbdf5eef60ed6b1199c0e8ee31ef78CAS |
[484] E. Setijadi, L. Tao, J. Liu, Z. Jia, C. Boyer, T. P. Davis, Biomacromolecules 2009, 10, 2699.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpsFGisb0%3D&md5=564eee15fd740be6b6bcde5ab835f265CAS |
[485] J. Liu, L. Tao, J. Xu, Z. Jia, C. Boyer, T. P. Davis, Polymer 2009, 50, 4455.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVOju7fJ&md5=a40f7f9f25c3b5b1c8f3810a6b0462faCAS |
[486] J. Skey, H. Willcock, M. Lammens, F. Du Prez, R. K. O’Reilly, Macromolecules 2010, 43, 5949.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXot1Gitr8%3D&md5=fd5cd948327ce39d145da701c166756cCAS |
[487] G. Johnston-Hall, M. J. Monteiro, Aust. J. Chem. 2009, 62, 857.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpvFeltb8%3D&md5=6759c1841abf250d2906036360ec6a52CAS |
[488] M. G. Fröhlich, M. M. Nardai, N. Förster, P. Vana, G. Zifferer, Polymer 2010, 51, 5122.
| Crossref | GoogleScholarGoogle Scholar |
[489] Y. Yan, W. Zhang, Y. Qiu, Z. Zhang, J. Zhu, Z. Cheng, W. Zhang, X. Zhu, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 5206.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVKjs7%2FN&md5=7780fe71694846d686747d2ea7f1d0aaCAS |
[490] Y. Gu, J. He, C. Li, C. Zhou, S. Song, Y. Yang, Macromolecules 2010, 43, 4500.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlt1Wrtr8%3D&md5=57832d03af3a7ad32c10f508e7b39cb1CAS |
[491] J. Song, R. Yu, L. Wang, S. Zheng, X. Li, Polymer 2011, 52, 2340.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltFentrc%3D&md5=4cc137cdb052de3056a1876b5c537fe5CAS |
[492] B. Quiclet-Sire, G. Revol, S. Z. Zard, Tetrahedron 2010, 66, 6656.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptlaqtLo%3D&md5=b9378c70b613665d2a880865a69415d3CAS |
[493] V. Patel, A. Mishra, N. Vishwakarma, C. Biswas, B. Ray, Polym. Bull. 2010, 65, 97.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkvFOqs74%3D&md5=682a5843e440abdcc89b60d26b786c89CAS |
[494] E. Girard, T. Tassaing, J.-D. Marty, M. Destarac, Polym. Chem. 2011, 2, 2222.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1eis7%2FF&md5=82c2cc1d30f1f8a13b68e2cff82da7abCAS |
[495] M. P. F. Pepels, C. I. Holdsworth, S. Pascual, M. J. Monteiro, Macromolecules 2010, 43, 7565.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVelurnK&md5=ccca0ba9cb5511cf9c474ab88de3d5ddCAS |
[496] W. Y. Tam, C. S. K. Mak, A. M. C. Ng, A. B. Djurisic, W. K. Chan, Macromol. Rapid Commun. 2009, 30, 622.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltl2gtLs%3D&md5=cc81e04457954480e49010761a1f499dCAS |
[497] A. K. Mishra, V. K. Patel, N. K. Vishwakarma, C. S. Biswas, M. Raula, A. Misra, T. K. Mandal, B. Ray, Macromolecules 2011, 44, 2465.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvFejsrg%3D&md5=07fef3ea53426723a505b6a581b8b9f2CAS |
[498] N. Fandrich, J. Falkenhagen, S. M. Weidner, B. Staal, A. F. Thünemann, A. Laschewsky, Macromol. Chem. Phys. 2010, 211, 1678.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXps1GhsL0%3D&md5=1f942c63e144a055d0fcbb5312e5efedCAS |
[499] M. Beija, E. Palleau, S. Sistach, X. Zhao, L. Ressier, C. Mingotaud, M. Destarac, J.-D. Marty, J. Mater. Chem. 2010, 20, 9433.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlSmt73L&md5=6050e80281f1a12dab483920ffdf7b68CAS |
[500] J. M. O’Donnell, E. W. Kaler, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 604.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1WisbjJ&md5=d6b4a9710fd01fcb19f0f35d6ef8edf6CAS |
[501] J. O’Donnell, E. W. Kaler, Macromolecules 2010, 43, 1730.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVSiu74%3D&md5=cdb1219d901dd99306cee7cc9ca57243CAS |
[502] S. A. Willis, G. R. Dennis, G. Zheng, W. S. Price, J. Mol. Liq. 2010, 156, 45.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFWiurnO&md5=ae5111884a71159276f42dab1970de93CAS |
[503] C. E. Lipscomb, M. K. Mahanthappa, Macromolecules 2009, 42, 4571.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmvVCis7c%3D&md5=8f22f12d6d2d7178bccd250d30c05c89CAS |
[504] E. Girard, J.-D. Marty, B. Ameduri, M. Destarac, ACS Macro Letters 2012, 1, 270.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmsVSnuw%3D%3D&md5=32bf6feb2d79c4f64d62fcb51a46b6c3CAS |
[505] M. Beija, J.-D. Marty, M. Destarac, Chem. Commun. 2011, 47, 2826.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXit1ejtr0%3D&md5=cf26ebf271abd82afc5c9eb9422566baCAS |
[506] M. Destarac, I. Blidi, O. Coutelier, A. Guinaudeau, S. Mazières, E. van Gramberen, J. Wilson, in “Progress in Controlled Radical Polymerization: Mechanisms and Techniques”, ACS Symposium Series Vol. 1100 2012, p. 259 (Eds K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky) (American Chemical Society: Washington, D.C.)
[507] A. Guinaudeau, S. Mazieres, D. J. Wilson, M. Destarac, Polym. Chem. 2012, 3, 81.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFGjsbrP&md5=53d7f31159f9a85ff014bef1ff406ec2CAS |
[508] I. Blidi, R. Geagea, O. Coutelier, S. Mazieres, F. Violleau, M. Destarac, Polym. Chem. 2012, 3, 609.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvFalurc%3D&md5=c53effa181415cf5a4a3368e554c3352CAS |
[509] M. Schellekens, D. Twene, A. van der Waals, Prog. Org. Coat. 2011, 72, 138.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpsF2jt7Y%3D&md5=8608c7abb3e8280fb7adfc6dde511abdCAS |
[510] I. S. Altarawneh, V. G. Gomes, M. H. Srour, J. Appl. Polym. Sci. 2009, 114, 2356.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVCgtbnM&md5=295fa43f36d70c1619b67dbe47817274CAS |
[511] I. S. Altarawneh, V. G. Gomes, M. H. Srour, Macromol. React. Eng. 2012, 6, 8.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFeqt7nP&md5=0540385a1f886e5adce5160aee662c10CAS |
[512] J. Schmitt, N. Blanchard, J. Poly, Polym. Chem. 2011, 2, 2231.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1eis7zN&md5=ac2e8f941034d657fcc8d52bcee665a2CAS |
[513] N. A. Birkin, N. J. Arrowsmith, E. J. Park, A. P. Richez, S. M. Howdle, Polym. Chem. 2011, 2, 1293.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnt1Kgs7k%3D&md5=959bd7e76c7b70116dc5863e914c9570CAS |
[514] N. Hu, W.-X. Ji, Y.-Y. Tong, Z.-C. Li, E.-Q. Chen, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 4621.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFGgsbbE&md5=83606990f9243f048bd470a9a8b44bd3CAS |
[515] C. F. Huang, J. A. Yoon, K. Matyjaszewski, Can. J. Chem. 2010, 88, 228.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhvFGmsLw%3D&md5=ec69a87f7935198a4fd90d58de20b2d1CAS |
[516] N. S. Ieong, M. Redhead, C. Bosquillon, C. Alexander, M. Kelland, R. K. O’Reilly, Macromolecules 2011, 44, 886.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1egsbs%3D&md5=57f6c50ac25cfd3b183b4bb5bb785c59CAS |
[517] Y. Maki, H. Mori, T. Endo, Macromol. Chem. Phys. 2010, 211, 1137.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmsFyqtLs%3D&md5=c369ce493d0f3c7093472c100f6badf5CAS |
[518] C. Xiao, D. Lu, S. Xu, L. Huang, Starch 2011, 63, 209.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkt1yqtLk%3D&md5=4c15ed997837e9c2bc5eed3e9b959370CAS |
[519] Y. Yu, H. Kang, J. Youk, Coll. Polym. Sci. 2012, 290, 1107.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XktFOrsL0%3D&md5=f622552c5555873c7e4dfedb55123201CAS |
[520] B. E. B. Jensen, A. A. A. Smith, B. Fejerskov, A. Postma, P. Senn, E. Reimhult, M. Pla-Roca, L. Isa, D. S. Sutherland, B. Städler, A. N. Zelikin, Langmuir 2011, 27, 10216.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovFKlsL0%3D&md5=36536247234986a3bced9b7ddc50c216CAS |
[521] A. A. A. Smith, T. Hussmann, J. Elich, A. Postma, M.-H. Alves, A. N. Zelikin, Polym. Chem. 2012, 3, 85.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFGjsb3M&md5=33127fd4eea48ddf7ae3f5ccf52f1de4CAS |
[522] X. Xue, J. Zhu, Z. Zhang, Z. Cheng, Y. Tu, X. Zhu, Polymer 2010, 51, 3083.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnvFWgsL8%3D&md5=2bc1292572887aada14b149a8d08114cCAS |
[523] D. Roy, B. S. Sumerlin, Polymer 2011, 52, 3038.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnsVSgt7g%3D&md5=5303e117ce5e223d55fcaef5a0cb9096CAS |
[524] J. Bernard, F. Lortie, B. Fenet, Macromol. Rapid Commun. 2009, 30, 83.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsleksLc%3D&md5=695ba2a7e63786eb5623aceab2fb75b7CAS |
[525] C.-F. Huang, R. Nicolay, Y. Kwak, F.-C. Chang, K. Matyjaszewski, Macromolecules 2009, 42, 8198.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVKksLfM&md5=b9789c61c6750b00db1b1280b277238eCAS |
[526] K. Chen, N. Grant, L. Liang, H. Zhang, B. Tan, Macromolecules 2010, 43, 9355.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtleitLbK&md5=4c4558d95a0572ec6ba6a31becfff489CAS |
[527] C. E. Lipscomb, M. K. Mahanthappa, Macromolecules 2011, 44, 4401.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlslKisr8%3D&md5=e0a058addecbb848567a19a91835c031CAS |
[528] Y. Qiu, W. Zhang, Y. Yan, J. Zhu, Z. Zhang, X. Zhu, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 5180.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVKjs77E&md5=1eb593e694c2b121787cebb9c534fec4CAS |
[529] C. M. R. Abreu, P. V. Mendonça, A. C. Serra, J. F. J. Coelho, A. V. Popov, G. Gryn’ova, M. L. Coote, T. Guliashvili, Macromolecules 2012, 45, 2200.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XislaksLc%3D&md5=15504fec079702d37702913e84dc490dCAS |
[530] M. Benaglia, M. Chen, Y. K. Chong, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 2009, 42, 9384.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVWhtr3N&md5=337ba2180bc3d4971308680740ad186aCAS |
[531] G. Moad, M. Benaglia, M. Chen, J. Chiefari, Y. K. Chong, D. J. Keddie, E. Rizzardo, S. H. Thang, in “Non-Conventional Functional Block Copolymers”, ACS Symposium Series Vol. 1066 2011, p. 81 (Eds P. Theato, A. F. M. Kilbinger, E. B. Coughlin) (American Chemical Society: Washington, D.C.).
[532] D. J. Keddie, C. Guerrero-Sanchez, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 2011, 44, 6738.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVags7nK&md5=daabb09aa1e88a4e12a702f889ceb83bCAS |
[533] U. Hasegawa, A. J. van der Vlies, E. Simeoni, C. Wandrey, J. A. Hubbell, J. Am. Chem. Soc. 2010, 132, 18273.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFSqsbnL&md5=3bef49b6c0121be210a76c2dbcc1b37dCAS |
[534] P. H. Hoang, C. T. Nguyen, J. Perumal, D.-P. Kim, Lab Chip 2011, 11, 329.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsF2jtb7F&md5=4e7fd54227599ce1beb8f4d9cc0d5b93CAS |
[535] C. T. Nguyen, Q. D. Nghiem, D.-P. Kim, J. San Chang, Y. K. Hwang, Polymer 2009, 50, 5037.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1antrvJ&md5=fa2ddb0129084556c6d16e97989ef8bcCAS |
[536] A. Nagai, R. Yoshii, T. Otsuka, K. Kokado, Y. Chujo, Langmuir 2010, 26, 15644.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFSlsLrN&md5=242e384892da3d0ed49046ed8ad0474eCAS |
[537] J. Zhang, R. Guo, G. Qi, M. Liu, A. Dong, Acta Polym. Sinica 2010, 010, 691.
| Crossref | GoogleScholarGoogle Scholar |
[538] J. Mao, X. Qi, X. Cao, J. Lu, Q. Xu, H. Gu, Chem. Commun. 2011, 47, 4228.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjs1Wlsr4%3D&md5=ea8ccd781651c505c0d4daaaa10aa3dcCAS |
[539] J. Qin, L. Zhang, H. Jiang, J. Zhu, Z. Zhang, W. Zhang, N. Zhou, Z. Cheng, X. Zhu, Chem.–Eur. J. 2012, 18, 6015.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XltVeisb4%3D&md5=2bb9110dcaeb0e2ce02a10a2da6349a6CAS |
[540] D. Hua, J. Tang, J. Jiang, X. Zhu, Polymer 2009, 50, 5701.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVWnurjM&md5=f22bdba6a06b0c7a6b2770691b6ac86aCAS |
[541] D. Hua, J. Tang, J. Jiang, X. Zhu, R. Bai, Macromolecules 2009, 42, 8697.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlSls7vE&md5=76fa76cb31a27fdcfe608e8486853313CAS |
[542] S. Jana, A. Parthiban, Macromol. Chem. Phys. 2011, 212, 790.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXksVehsb4%3D&md5=68f41ef5f23bae3fd97159b559ee3153CAS |
[543] A. Lubnin, K. O’Malley, D. Hanshumaker, J. Lai, Eur. Polym. J. 2010, 46, 1563.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnvFWmt7o%3D&md5=0751837606d2239ff44b22d8f528fb8cCAS |
[544] Q. D. Nghiem, C. T. Nguyen, D.-P. Kim, J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 4594.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXot1ehtb4%3D&md5=a55cddc8afffcf2139957d59cd6f7e3bCAS |
[545] R. Skouta, S. Wei, R. Breslow, J. Am. Chem. Soc. 2009, 131, 15604.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1GgsLjE&md5=db55d5ba40d64adf43f0acbd7a62ff32CAS |
[546] L. Nebhani, S. Sinnwell, C. Y. Lin, M. L. Coote, M. H. Stenzel, C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 6053.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1OhtbzL&md5=7d799c277823d26e3b5b836887f351ecCAS |
[547] L. Nebhani, S. Sinnwell, A. J. Inglis, M. H. Stenzel, C. Barner-Kowollik, L. Barner, Macromol. Rapid Commun. 2008, 29, 1431.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtF2jt7jE&md5=79969a0e7262c28fcac23c9c200e3a0fCAS |
[548] J. Zhou, N. K. Guimard, A. J. Inglis, M. Namazian, C. Y. Lin, M. L. Coote, E. Spyrou, S. Hilf, F. G. Schmidt, C. Barner-Kowollik, Polym. Chem. 2012, 3, 628.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvFaksrc%3D&md5=937bfc34e5bcc7df4c63e36491b290edCAS |
[549] R. Geagea, S. Ladeira, S. Mazières, M. Destarac, Chem.–Eur. J. 2011, 17, 3718.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjtFWnsbo%3D&md5=f70939037d68854e955915e8ea887b49CAS |
[550] G. Moad, E. Rizzardo, S. H. Thang, Acc. Chem. Res. 2008, 41, 1133.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXpvVOqu78%3D&md5=587ee23e42e0e746d7cc4ba808d4d986CAS |
[551] E. Rizzardo, G. Moad, S. H. Thang, Reversible Addition Fragmentation Chain Transfer, in Encyclopedia of Polymer Science and Technology 2009 (John Wiley & Sons, Inc.: Hoboken, NJ).
[552] J. Skey, R. K. O’Reilly, Chem. Commun. 2008, 4183.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFektbzL&md5=d76711c0cab0dd92c4be07afdcdc9aabCAS |
[553] F. Lebreux, B. Quiclet-Sire, S. Z. Zard, Org. Lett. 2009, 11, 2844.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXntVSrsr8%3D&md5=69ea804288c936f4ee96611f80edea16CAS |
[554] K. Skrabania, A. Miasnikova, A. M. Bivigou-Koumba, D. Zehm, A. Laschewsky, Polym. Chem. 2011, 2, 2074.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVOltbbF&md5=ee5e8acfa0b74651422b9ccd6a53e0b6CAS |
[555] S. Xiao, P. A. Charpentier, Acta Crystallogr. E 2011, 67, o811.
[556] T. Gruendling, S. Weidner, J. Falkenhagen, C. Barner-Kowollik, Polym. Chem. 2010, 1, 599.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlOhtb7K&md5=0b5e9293964f02438d4360d863a78bc5CAS |
[557] A. Baumgaertel, E. Altuntaş, U. S. Schubert, J. Chromatogr. A 2012, 1240, 1.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xms1yku7g%3D&md5=672a2a4f5e82a4455660d4adf9d78393CAS |
[558] D. Berek, Anal. Bioanal. Chem. 2010, 396, 421.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtl2rt7fN&md5=8b7996cdf483652f767c5e42370c658cCAS |
[559] D. Pissuwan, C. Boyer, K. Gunasekaran, T. P. Davis, V. Bulmus, Biomacromolecules 2010, 11, 412.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlvFWjtA%3D%3D&md5=bb4f7614500febcfad3808f2e4ea3d75CAS |
[560] J. C. Hernández-Ortiz, G. Jaramillo-Soto, J. Palacios-Alquisira, E. Vivaldo-Lima, Macromol. React. Eng. 2010, 4, 210.
| Crossref | GoogleScholarGoogle Scholar |
[561] B. Bitsch, C. Barner-Kowollik, S. Zhu, Macromol. React. Eng. 2011, 5, 55.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXit12itg%3D%3D&md5=c5a68e53900c846bc0db0b02a580c1b2CAS |
[562] A. M. Alb, W. F. Reed, Macromol. React. Eng. 2010, 4, 470.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpvFKhu78%3D&md5=18688e548116fe7c00649d0d8f829878CAS |
[563] Y. Wu, Q. Wang, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 2425.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlsVKmsL8%3D&md5=a9f78f3fc9279a982990cfde018cd2e5CAS |
[564] R. M. Paulus, C. R. Becer, R. Hoogenboom, U. S. Schubert, Aust. J. Chem. 2009, 62, 254.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjsVKqtrY%3D&md5=a8abb041acae301c9513692d15ff58b5CAS |
[565] P. B. Zetterlund, S. b. Perrier, Macromolecules 2011, 44, 1340.
| 1:CAS:528:DC%2BC3MXitVShsLg%3D&md5=86a227f88c803d808d43a452e8ac93bbCAS |
[566] C. H. Hornung, A. Postma, S. Saubern, J. Chiefari, Macromol. React. Eng. 2012, 6, 246.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XovFWluro%3D&md5=f0d5b50d1c9c6bc6d98b01c67f243450CAS |
[567] A. Zargar, F. J. Schork, Ind. Eng. Chem. Res. 2009, 48, 4245.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjvVOmurw%3D&md5=c915402fcbfd0b7b842549bdf91b2481CAS |
[568] J. Bernard, M. Save, B. Arathoon, B. Charleux, J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2845.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXltVOiu74%3D&md5=a44f493bbd7e28ffe31dbe1e76cb9f11CAS |
[569] Z. Jia, M. J. Monteiro, in “Progress in Controlled Radical Polymerization: Mechanisms and Techniques”, ACS Symposium Series Vol. 1100 2012, p. 293 (Eds K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky) (American Chemical Society: Washington, D.C.)
[570] A. Sogabe, C. L. McCormick, Macromolecules 2009, 42, 5043.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotVGksbs%3D&md5=2a062c65fa6e2530108cececdb219ee3CAS |
[571] C. Grazon, J. Rieger, N. Sanson, B. Charleux, Soft Matter 2011, 7, 3482.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsFylsL4%3D&md5=bb9523e7dc7d965aa51ce9fd04ac2668CAS |
[572] R. K. O’Reilly, Polym. Int. 2010, 59, 568.
| 1:CAS:528:DC%2BC3cXlt1CitL0%3D&md5=d7472f38363da56bfc89676b2306c3d2CAS |
[573] M. Destarac, D. Taton, S. Z. Zard, T. Saleh, Y. Six, in “Advances in Controlled/Living Radical Polymerization”, ACS Symposium Series Vol. 854 2003, p. 536 (Ed. K. Matyjaszewski) (American Chemical Society: Washington, D.C.).
[574] Y. Reyes, J. M. Asua, Macromol. Rapid Commun. 2011, 32, 63.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1Wqu73L&md5=2f0145595ea01fba5455dec14cef0476CAS |
[575] M. Gaborieau, S. P. S. Koo, P. Castignolles, T. Junkers, C. Barner-Kowollik, Macromolecules 2010, 43, 5492.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnsVSrtb8%3D&md5=4b6910a24d862bbf05629a6e4f3bba99CAS |
[576] K. Liang, R. A. Hutchinson, J. Barth, S. Samrock, M. Buback, Macromolecules 2011, 44, 5843.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovVOjsbo%3D&md5=e773cc3c94446e39b9357549b15e1833CAS |
[577] M. Turson, M. Zhou, P. Jiang, X. Dong, J. Sep. Sci. 2011, 34, 127.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosVCjtg%3D%3D&md5=be2de60d5d0878c135aca905b7ce0368CAS |
[578] A. Nagai, K. Kokado, J. Miyake, Y. Chujo, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 627.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1WisbjF&md5=8f54aa3368cecde5fda2976af39ad187CAS |
[579] A. Nagai, K. Kokado, J. Miyake, Y. Chujo, Macromolecules 2009, 42, 5446.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotF2ktr8%3D&md5=1916c68608f76b9a5dfa1df64c34dee1CAS |
[580] Y. Zhou, S.-k. Ahn, R. K. Lakhman, M. Gopinadhan, C. O. Osuji, R. M. Kasi, Macromolecules 2011, 44, 3924.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltFGmtLY%3D&md5=3fe56896ddfb0c7967a64b47ab827c1bCAS |
[581] H.-S. Sohn, S.-H. Cha, W.-K. Lee, D.-G. Kim, H.-J. Yun, M.-S. Kim, B.-D. Kim, Y.-H. Kim, J.-W. Lee, J.-S. Kim, D.-B. Kim, J.-H. Kim, J.-C. Lee, Macromol. Res. 2011, 19, 722.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXot1Cks78%3D&md5=43c27dfb28ab4b1d5b72acdfcd203779CAS |
[582] X. Xu, A. E. Smith, C. L. McCormick, Aust. J. Chem. 2009, 62, 1520.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVers7nL&md5=4ef21c49f01360e5153a1370a7896a39CAS |
[583] F. Cheng, F. Jakle, Chem. Commun. 2010, 46, 3717.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmtFKqu7g%3D&md5=eb801629ac1b5e0924d5da3836bc743eCAS |
[584] M. Semsarilar, V. Ladmiral, S. Perrier, Macromolecules 2010, 43, 1438.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnvFSh&md5=6f32d37b6fe81f27e417fb813c0f0beaCAS |
[585] R. Kakuchi, P. Theato, Macromolecules 2012, 45, 1331.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsVyks7w%3D&md5=071d525590db795dfbd906a0babdac31CAS |
[586] J.-J. Yan, C.-Y. Hong, Y.-Z. You, Macromolecules 2011, 44, 1247.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVekur0%3D&md5=7052ac8751b43411ea56aa146f2a38f0CAS |
[587] M. Seo, S. Shin, S. Ku, S. Jin, J.-B. Kim, M. Ree, S. Y. Kim, J. Mater. Chem. 2010, 20, 94.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFahur7N&md5=c49a46f03b883ffe00741553b06fffcdCAS |
[588] J. A. Syrett, D. M. Haddleton, M. R. Whittaker, T. P. Davis, C. Boyer, Chem. Commun. 2011, 47, 1449.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXot1Crtw%3D%3D&md5=e886b5a94deb6a0fd55cf6bd5bb46b54CAS |
[589] S. Erkoc, A. E. Acar, Macromolecules 2008, 41, 9019.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlemt7zO&md5=2acb32e83bb3c83b8744fca2983312f1CAS |
[590] H. Mori, M. Tsukamoto, Polymer 2011, 52, 635.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlKrurg%3D&md5=9a22e05fd1e696a63d164958871e2b04CAS |
[591] B. Ochiai, Y. Ootani, T. Endo, J. Am. Chem. Soc. 2008, 130, 10832.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXovFygu7k%3D&md5=05eedd2bae2ea72149d6a503560fb697CAS |
[592] A. K. Sharma, C. Cornaggia, D. Pasini, Macromol. Chem. Phys. 2010, 211, 2254.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVWgu77E&md5=44c9afc400d1409aca1a490f5d60dc88CAS |
[593] Y. K. Chong, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 2007, 40, 4446.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlsVKisb0%3D&md5=966471da00ec76ebc2948d026416c2ccCAS |
[594] T. Hou, P. Zhang, X. Zhou, X. Cao, Y. Zhao, Chem. Commun. 2010, 46, 7397.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1Snt7%2FJ&md5=db733290890cc44f86d8b79f1005c5a8CAS |
[595] C.-D. Vo, J. Rosselgong, S. P. Armes, N. Tirelli, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 2032.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjvV2qurY%3D&md5=3aec047a6ae188838453acc291059eb0CAS |
[596] L. Houillot, C. Bui, C. l. Farcet, C. Moire, J.-A. Raust, H. Pasch, M. Save, B. Charleux, ACS Appl. Mater. Interfaces 2010, 2, 434.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnt1Sgsw%3D%3D&md5=7d0adb5cb5cece467f3a1b1911cf29baCAS |
[597] B. Du, A. Mei, Y. Yang, Q. Zhang, Q. Wang, J. Xu, Z. Fan, Polymer 2010, 51, 3493.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXosVGmu7c%3D&md5=f16f762cc28c31a4b4eac847fdaafc46CAS |
[598] Y. Zhang, Z. Shen, D. Yang, C. Feng, J. Hu, G. Lu, X. Huang, Macromolecules 2010, 43, 117.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlKms7%2FE&md5=b6fd783d031362dc5d809172b81d33cbCAS |
[599] K. J. Thurecht, I. Blakey, H. Peng, O. Squires, S. Hsu, C. Alexander, A. K. Whittaker, J. Am. Chem. Soc. 2010, 132, 5336.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjvFKjt74%3D&md5=c34bf0bbc5f358cf9465f4f5c6630141CAS |
[600] F. D. Jochum, L. zur Borg, P. J. Roth, P. Theato, Macromolecules 2009, 42, 7854.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVOrs7zP&md5=b944c8bed442ec53f727179d9e6ae112CAS |
[601] P. Scheibe, M. Barz, M. Hemmelmann, R. Zentel, Langmuir 2010, 26, 5661.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjvFertbk%3D&md5=36333e0e0da77b1829cb0a483e5e29c8CAS |
[602] A. H. Soeriyadi, C. Boyer, J. Burns, C. R. Becer, M. R. Whittaker, D. M. Haddleton, T. P. Davis, Chem. Commun. 2010, 46, 6338.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVaqtrvN&md5=d94e3afffd30631046eb8ab22ea72834CAS |
[603] K. Yamamoto, A. Takasu, Macromolecules 2010, 43, 8519.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtF2ru7%2FJ&md5=a7b182c4694eaf1a520d4ef2531623e7CAS |
[604] B. Ebeling, P. Vana, Polymers 2011, 3, 719.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltF2rurY%3D&md5=b4ad645fb52f81f79c64e876d2fd1b46CAS |
[605] T. Gruendling, M. Dietrich, C. Barner-Kowollik, Aust. J. Chem. 2009, 62, 806.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpvFelt7o%3D&md5=431884d9b4e1a2d2b100f54802dd8dc2CAS |
[606] B. Quiclet-Sire, S. Z. Zard, Bull. Korean Chem. Soc. 2010, 31, 543.
| Crossref | GoogleScholarGoogle Scholar |
[607] S. Zard, B. Sire, P. Jost, US Patent 7 473 740 2009.
[608] R. Pfukwa, G. Pound, B. Klumperman, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 2008, 49, 117.
| 1:CAS:528:DC%2BD1cXpslShtbk%3D&md5=45391a561120f1fbd63834042001256aCAS |
[609] F. Grellepois, C. Portella, Synthesis 2008, 2008, 3443.
| Crossref | GoogleScholarGoogle Scholar |
[610] Y. Zhou, J. He, C. Li, L. Hong, Y. Yang, Macromolecules 2011, 44, 8446.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1KktrfN&md5=d90703136a2f69fc11a852680034dacbCAS |
[611] E. D. Pressly, R. J. Amir, C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 814.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1ejurzI&md5=d1e1fc3564b2d12d2a62f4936d7b9d8aCAS |
[612] G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer, D. M. Haddleton, Polym. Chem. 2010, 1, 1196.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtl2msLrO&md5=d809707c783f07ad2b4aecc834b609f4CAS |
[613] L. Nebhani, P. Gerstel, P. Atanasova, M. Bruns, C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 7090.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVymurzJ&md5=d8844b9ca159d25acf03bf5cff2ed71aCAS |
[614] A. Bousquet, C. Boyer, T. P. Davis, M. H. Stenzel, Polym. Chem. 2010, 1, 1186.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtl2msLrN&md5=16f5242f75770604db7d405844a37b9bCAS |
[615] A. J. Inglis, T. Paulöhrl, C. Barner-Kowollik, Macromolecules 2010, 43, 33.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFaksLvE&md5=bf789f0f5fdc37a05513595c5f2a93e1CAS |
[616] H. Dentel, I. Chataigner, F. Le Cavelier, M. Gulea, Tetrahedron Lett. 2010, 51, 6014.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlSmt7jI&md5=a684a4d1a3225a8b0ee6693865fd9e47CAS |
[617] T. Paulöhrl, A. J. Inglis, C. Barner-Kowollik, Adv. Mater. 2010, 22, 2788.
| Crossref | GoogleScholarGoogle Scholar |
[618] D. Pavlović, J. G. Linhardt, J. F. Künzler, D. A. Shipp, Macromol. Chem. Phys. 2010, 211, 1482.
| Crossref | GoogleScholarGoogle Scholar |
[619] A. Favier, B. Luneau, J. Vinas, N. Laissaoui, D. Gigmes, D. Bertin, Macromolecules 2009, 42, 5953.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXovVemtrY%3D&md5=8329069f026ecb5601900fd7037ea7eeCAS |
[620] C. Zhou, M. A. Hillmyer, T. P. Lodge, Macromolecules 2011, 44, 1635.
| 1:CAS:528:DC%2BC3MXisVWlu7g%3D&md5=e6ef3640a7293d3eda5e1fe86ada107bCAS |
[621] C. C. Williams, S. H. Thang, T. Hantke, U. Vogel, P. H. Seeberger, J. Tsanaktsidis, B. Lepenies, ChemMedChem 2012, 7, 281.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFKksrzF&md5=c793de61e17739b54b75e15fd179344dCAS |
[622] N. A. Hadjiantoniou, T. Krasia-Christoforou, E. Loizou, L. Porcar, C. S. Patrickios, Macromolecules 2010, 43, 2713.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhvFOrtr8%3D&md5=a7d2010de35f2c21f0f46aacf6082befCAS |
[623] B. Ebeling, M. Eggers, P. Vana, Macromolecules 2010, 43, 10283.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVGksbrP&md5=a42e2abb6afaeb3c5b43c46e19d35f90CAS |
[624] Y. Liu, K. A. Cavicchi, Macromol. Chem. Phys. 2009, 210, 1647.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1aqs77P&md5=65f0089d2234ad9e4d843eea320180d6CAS |
[625] S. Z. Luo, X. L. Hu, Y. Y. Zhang, C. X. Ling, X. Liu, S. S. Chen, Polym. J. 2011, 43, 41.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVCnug%3D%3D&md5=d4e2a40636b44794e54b72ec7e561c8cCAS |
[626] M. L. Koh, D. Konkolewicz, S. Perrier, Macromolecules 2011, 44, 2715.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXktFentL4%3D&md5=a7438da1ff35747dee40daec2d39fcafCAS |
[627] R. l. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu, H.-A. Klok, Chem. Rev. 2009, 109, 5437.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht12iurbK&md5=7b70db53cf2247d22292236afa6d15e1CAS |
[628] J. Bolton, J. Rzayev, ACS Macro Letters 2012, 1, 15.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVequr%2FI&md5=7a44f4ae22484517a66be67ce2a8557eCAS |
[629] M. Semsarilar, V. Ladmiral, S. Perrier, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 4361.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVyiu7%2FO&md5=017307519885f19a984a04887152ceb7CAS |
[630] D. Wu, X. Song, T. Tang, H. Zhao, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 443.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFOqtbvN&md5=738e4a32d56a608e076a5b0081518fafCAS |
[631] A. Nese, Y. Kwak, R. Nicolaÿ, M. Barrett, S. S. Sheiko, K. Matyjaszewski, Macromolecules 2010, 43, 4016.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkvV2rsr8%3D&md5=6d8796ccbee0f8ce126f726f4836f8a1CAS |
[632] J. Zhou, W. Li, J.-S. Gu, H.-Y. Yu, Z.-Q. Tang, X.-W. Wei, Sep. Pur. Technol. 2010, 71, 233.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtleitr0%3D&md5=8e196fc0f821e16a2aece59dfa9f6c9dCAS |
[633] B. Zhang, Y. Chen, L. Xu, L. Zeng, Y. He, E.-T. Kang, J. Zhang, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 2043.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsFCmtr0%3D&md5=81c46d6dfa83079e70957fe56a8d9113CAS |
[634] C. Grande, M. Tria, M. Felipe, F. Zuluaga, R. Advincula, Eur. Phys. J. E 2011, 34, 15.
| Crossref | GoogleScholarGoogle Scholar |
[635] S. Z. Guo, C. Zhang, W. Z. Wang, T. X. Liu, Express Polym. Lett. 2010, 4, 17.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlvF2q&md5=f7136e48e0dda0aa59f1b8541301732eCAS |
[636] M. Turson, X. L. Zhuang, H. N. Liu, P. Jiang, X. C. Dong, Chin. Chem. Lett. 2009, 20, 1136.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlOitLjF&md5=9deb40eef75d88afa22444c12aa341d4CAS |
[637] T. P. Le, G. Moad, E. Rizzardo, S.H. Thang, US Patent 7 666 962 2010.
[638] T. P. Le, G. Moad, E. Rizzardo, S. H. Thang, US Patent 7 662 986 2010.
[639] T. P. Le, G. Moad, E. Rizzardo, S. H. Thang, US Patent 7 714 075 2010.
[640] R. Padiyath, M. D. Radcliffe, C. U. Thomas, C. A. Marttila, M. Jain, M. F. Weber, F. Bai, US Patent 7 652 736 2010.
[641] G. Moad, G. Li, J. A. Campbell, H. Wermter, R. Pfaendner, US Patent 8 110 626 2012.
[642] G. Moad, G.P. Simon, K.M. Dean, G. Li, R.T.A. Mayadunne, H. Wermter, R. Pfaendner, US Patent 7 837 899 2010.
[643] F. Louwet, G. Deroover, B. Groenendaal, M. Hartenstein, J. Storsberg, US Patent 7 674 843 2010.
[644] F. Louwet, G. Deroover, B. Groenendaal, M. Hartenstein, J. Storsberg, US Patent 7 678 845 2010.
[645] B. Groenendaal, G. Deroover, US Patent 7 691 937 2010.
[646] S. A. Curran, A. V. Ellis, US Patent 7 713 508 2010.
[647] J. G. Linhardt, J. A. McGee, I. M. Nunez, US Patent 8 133 960 2012.
[648] J. G. Linhardt, D. A. Shipp, J. F. Kunzler, D. P. Vanderbilt, US Patent 8 100 528 2012.
[649] J. G. Linhardt, J. F. Kunzler, D. A. Shipp, US Patent 8 083 348 2011.
[650] J. G. Linhardt, I. M. Nunez, J. A. McGee, J. Hunt, M. Alton, D. Pavlovic, D. A. Shipp, US 8 043 369 2011.
[651] J. G. Linhardt, D. A. Shipp, J. F. Kunzler, US Patent 7 942 929 2011.
[652] M.-T. Charreyre, B. Mandrand, J. M. G. Martinho, P. Relogio, J. P. Sequeira Farinha, US Patent 8 133 411 2012.
[653] R. E. Richard, US Patent 7 517 914 2009.
[654] B. Göbelt, J. Omeis, S. Johann, US Patent 7 851 544 2010.
[655] K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky, J. Spanswick, US Patent 7 795 355 2010.
[656] Y. Kensicher, D. Platel, US Patent 8 110 130 2012.
[657] J.-M. Suau, C. Jacquemet, US Patent 8 053 497 2011.
[658] J.-M. Suau, C. Jacquemet, US Patent 7 956 211 2011.
[659] J.-M. Suau, C. Jacquemet, US Patent 7 851 572 2010.
[660] C. Cheng, E. Khoshdel, K. L. Wooley, US Patent 7 960 479 2011.
[661] J. Z. Zhao, US Patent 7 811 555 2010.
[662] L. Meagher, H. Thissen, P. Pasic, R. A. Evans, G. Johnson, US Patent 8 124 188 2012.
[663] W. B. Farnham, G. Moad, S. H. Thang, E. Rizzardo, M. Fryd, US Patent 7 807 755 2010.
[664] W. B. Farnham, M. Fryd, F. L. Schadt, US Patent 7 696 292 2010.
[665] W. B. Farnham, S. Moudgil, US Patent 8 034 534 2011.
[666] W. B. Farnham, US Patent 7 632 966 2009.
[667] K. Mizutani, S. Sugiyama, US Patent 8 142 977 2012.
[668] D. K. Parker, F. J. Feher, V. Mahadevan, US Patent 8 178 637 2012.
[669] D. K. Parker, F. J. Feher, V. Mahadevan, US Patent 7 812 108 2010.
[670] D. K. Parker, J. J. Kulig, US Patent 7 671 152 2010.
[671] D. K. Parker, F. J. Feher, V. Mahadevan, US Patent 7 767 775 2010.
[672] D. K. Parker, X. Yang, US Patent 7 625 985 2009.
[673] D. K. Parker, F. J. Feher, V. Mahadevan, US Patent 7 592 409 2009.
[674] D. K. Parker, F. J. Feher, V. Mahadevan, US Patent 7 528 204 2009.
[675] J. Cheng, H.-C. Kim, C. T. Rettner, D. P. Sanders, R. Sooriyakumaran, L. Sundberg, US Patent 7 989 026 2011.
[676] J. Cheng, H.-C. Kim, D. P. Sanders, L. Sundberg, US Patent 7 763 319 2010.
[677] J. Cheng, W. D. Hinsberg, H.-C. Kim, C. T. Rettner, D. P. Sanders, US Patent 7 521 094 2009.
[678] J. Cheng, H. -C. Kim, C. T. Rettner, D. P. Sanders, R. Sooriyakumaran, L. Sundberg, US Patent 7 521 090 2009.
[679] K. Breitenkamp, K. Sill, H. Skaff, US Patent 7 612 153 2009.
[680] K. C. Shih, P. W. Chung, M. H. Wang, US Patent 7 968 743 2011.
[681] J. H. K. Charles, G. M. Murray, US Patent 7 799 568 2010.
[682] G. E. Southard, G. M. Murray, US Patent 7 678 870 2010.
[683] D. Benoit, A. Safir, H. -T. Chang, D. Charmot, I. Nishimura, A. Soyano, K. Okamoto, Y. Wang, , US Patent 7 510 817 2009.
[684] D. Benoit, A. Safir, H. -T. Chang, D. Charmot, K. Okamoto, I. Nishimura, Y. Wang, US Patent 7 517 634 2009.
[685] T. J. Lynch, US Patent 7 875 213 2011.
[686] D. Achten, M. Klimpel, E. Barriau, L. Reif, R. Mottweiler, H. Berg, Z. Szentivanyi, S. Glander, US Patent 7 825 193 2010.
[687] N. P. Boupat, O. Guerret, N. Mougin, X. Schultze, F. Hernandez, US Patent 7 951 888 2011.
[688] C. Farcet, US Patent 7 816 464 2010.
[689] N. P. Boupat, O. Guerret, N. Mougin, X. Schultze, F. Hernandez, US Patent 7 632 905 2009.
[690] C. Farcet, US Patent 7 585 922 2009.
[691] J. T.-Y. Lai, A. D. Pajerski, R. P. Shea, US Patent 8 137 754 2012.
[692] J. R. Johnson, D. C. Visger, M. Baum, B. J. Schober, Y. Wang, US Patent 8 012 917 2011.
[693] J. T.-Y. Lai, US Patent 7 851 582 2010.
[694] J. T.-Y. Lai, US Patent 7 659 345 2010.
[695] J. T.-Y. Lai, US Patent 7 495 128 2009.
[696] J. T.-Y. Lai, S.-J. R. Hsu, US Patent 7 495 050 2009.
[697] J. T.-Y. Lai, US Patent 7 498 456 2009.
[698] L. A. Hough, G. M. Lizarraga, H. Adam, J.-C. Castaing, S. Kesavan, US Patent 7 789 160 2010.
[699] S. Zard, B. Sire, P. Jost, US Patent 7 473 740 2009.
[700] N. Cadena, M. Destarac, P. Herve, A. Z. Wilczewska, US Patent 7 473 730 2009.
[701] P. Herve, M. Destarac, O. Anthony, B. Bavouzet, M. Joanicot, A. Z. Wilczewska, US Patent 7 531 597 2009.
[702] R. K. Prud'homme, I. A. Aksay, D. Adamson, US Patent 7 659 350 2010.
[703] J. Di, B. Mullen, P. D. Sybert, US Patent 7 498 398 2009.
[704] T. Dollase, M. Husemann, B. Luhmann, US Patent 8 129 470 2012.
[705] M. Husemann, S. Zollner, US Patent 8 012 581 2011.
[706] T. Dollase, M. Husemann, US Patent 7 758 933 2010.
[707] T. Dollase, M. Koop, US Patent 7 605 212 2009.
[708] M. Husemann, S. Zollner, US Patent 7 514 515 2009.
[709] S. Hansen, M. Husemann, K. Brandes, S. Zollner, US Patent 7 521 487 2009.
[710] C. J. Hawker, R. Vestberg, N. Ueno, US Patent 8 153 729 2012.
[711] C. N. Bowman, T. F. Scott, US Patent 7 943 680 2011.
[712] S. Thayumanavan, US Patent 7 687 600 2010.
[713] C. D. Frisbie, T. P. Lodge, US Patent 7 999 020 2011.
[714] C. L. McCormick, A. B. Lowe, B. S. Sumerlin, US Patent 8 084 558 2011.
[715] C. W. Scales, F. Huang, C. L. McCormick, US Patent 7 718 432 2010.
[716] C. H. Such, E. Rizzardo, A. K. Serelis, B. S. Hawkett, R. G. Gilbert, C. J. Ferguson, R. J. Hughes, l.r.E. Olejnik, US Patent 7 745 553 2010.
[717] P. S. Stayton, A. S. Hoffman, J.-i. Lai, J. Hoffman, M. Ebara, US Patent 7 981 688 2011.
[718] P. S. Stayton, A. S. Hoffman, X. Yin, L. J. Taite, J. Garbern, US Patent 7 718 193 2010.
[719] E. M. Harth, A. E. Van Der Ende, S. K. Hamilton, T. A. Croce, US Patent 7 935 782 2011.
[720] M. R. Linford, L. Yang, L. Wiest, US Patent 8 147 985 2012.
[721] R. G. Gilbert, M. Hess, A. D. Jenkins, R. G. Jones, P. Kratochvíl, R. F. T. Stepto, Pure Appl. Chem. 2009, 81, 351.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXisFGhsrY%3D&md5=bffb3f58ee98ff9e360e91ad6d97b25fCAS |
† For an explanation of the acronyms and abbreviations used throughout this paper please see the section entitled Abbreviations.