

The Evolving Coordination Chemistry of Radiometals for Targeted Alpha Therapy
Melyssa L. Grieve A and Brett M. Paterson A B C
A and Brett M. Paterson A B C
A School of Chemistry, Monash University, Clayton, Vic. 3800, Australia.
B Monash Biomedical Imaging, Monash University, Clayton, Vic. 3800, Australia.
C Corresponding author. Email: brett.paterson@monash.edu

Melyssa Grieve graduated from Monash University in 2019 with a Bachelor of Science – Advanced Research (Honours) majoring in genetics and chemistry. In 2020 she joined the group of Dr Brett Paterson at Monash University as a Ph.D. student working on developing chelators for use in radiopharmaceuticals. |

Dr Brett Paterson is a graduate of the University of Melbourne where Professor Paul Donnelly supervised his Ph.D. studies. This was followed by post-doctoral research with Professor Donnelly and a Victorian Post-Doctoral Fellowship at King's College London with Professor Phil Blower. Dr Paterson returned to Australia and received a Discovery Early Career Research Award from the Australian Research Council, which he undertook at Monash University. Dr Paterson was a National Imaging Facility Fellow at Monash Biomedical Imaging and is currently Head of Radiochemistry at the Centre for Advanced Imaging, University of Queensland. |
Australian Journal of Chemistry 75(2) 65-88 https://doi.org/10.1071/CH21184
Submitted: 2 August 2021 Accepted: 29 October 2021 Published: 7 December 2021
Journal Compilation © CSIRO 2022 Open Access CC BY
Abstract
Several radiometals are of interest in the development of new α-emitting radiopharmaceuticals. This review highlights the role of coordination chemistry in the design of 225Ac, 212/213Bi, 212Pb, 149Tb, 227Th, and 223/224Ra radiopharmaceuticals to treat cancer. Several chelators have recently been developed that are addressing the specific requirements of each radiometal to provide outstanding radiolabelling and in vivo properties. These advances are supporting the momentum that is building around radiopharmaceuticals for targeted α therapy.
Keywords: radiometals, targeted alpha therapy, coordination chemistry, radiopharmaceuticals, radiolabelling, chelators, inorganic chemistry, nuclear medicine.
Introduction
Radiopharmaceuticals are molecules that have been labelled with a radionuclide that emits ionising radiation to achieve non-invasive diagnostic imaging or deliver a therapeutic dose of ionising radiation to specifically targeted tissue. The α-particle is a nucleus of helium-4 (4He)2+, consisting of two protons and two neutrons. Owing to their high positive charge and heavy mass, α-particles are highly ionising with very high linear energy transfer (LET) resulting in energy deposition of 50–230 keV μm−1. The cytotoxic effect of therapeutic radionuclides is primarily due to their ability to induce irreversible DNA damage. The high kinetic energy (5–9 MeV) and short tissue range (50–100 μm, <10 cell diameters) of α-particles make them suitable for the treatment of small metastatic tumours or circulating malignant cells.
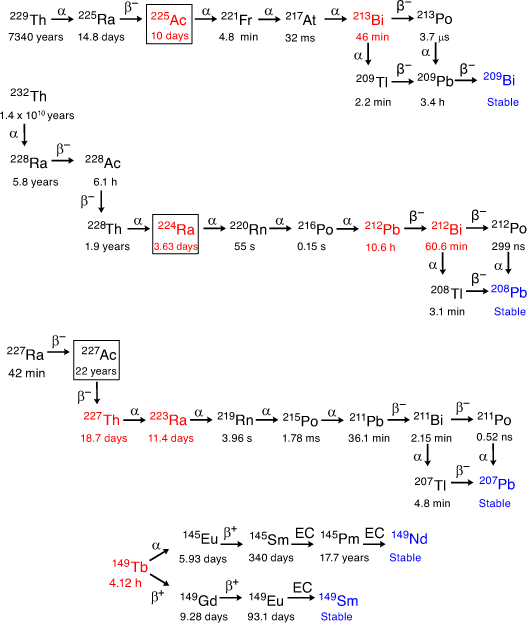
Although α-particles have been clinically demonstrated to be powerful tools for cancer therapy, there are several challenges that must be addressed before targeted α therapies (TAT) are regularly translated into the clinic. To be suitable for TAT, α-emitting radionuclides must have half-lives that are neither too short nor too long, and their production must be both economically viable and capable of widespread implementation. The most appropriate candidates have tended to be radionuclides of large metal ions. The most widely studied α-emitting radiometals to date are actinium-225 (225Ac, half-life t1/2 9.9 days), bismuth-213 (213Bi, t1/2 45.6 min), bismuth-212 (212Bi, t1/2 60.6 min), lead-212 (212Pb, t1/2 10.2 h), terbium-149 (149Tb, t1/2 4.1 h), thorium-227 (227Th, t1/2 18.7 days), radium-223 (223Ra, t1/2 11.4 days), and radium-224 (224Ra, t1/2 3.7 days) (Fig. 1). Currently, [223Ra][RaCl2] (Xofigo®) is the only clinically approved therapeutic α-emitting radiopharmaceutical with marketing approval, and is used in the treatment of metastatic castration-resistant prostate cancer (mCRPC).[1]
|
Viable chemistry must be developed to produce radiopharmaceuticals that treat the many and varied forms of cancer. Safe and effective delivery of radionuclides can be achieved with the use of coordination chemistry to produce complexes that effectively bind the radiometal ion. The small charge density of these large α-emitting radiometals requires distinct chelator design strategies. Opportunities exist to investigate various ligand donor atom preferences, coordination numbers, and coordination geometries. Chelators are evaluated based on the conditions (temperature, chelator concentration, pH, and reaction time) required to achieve near-quantitative radiolabelling efficiency and on the stability of the radiometal complexes to relevant in vivo conditions. Formation constants can be useful in preliminary evaluations but it is typically kinetic dissociation rates that govern in vivo stability. Competition experiments that involve biologically relevant ions or proteins can be used to assess the ability of the complexes to resist transchelation. Biodistribution in vivo is the most relevant and practical test of stability and to assess blood clearance and uptake profiles. Several chelators that have been investigated for binding α-emitting radiometals are shown in Fig. 2.

|
The fate of the daughter radionuclides after α decay is a general concern around TAT, particularly for multiple α-emitting radionuclides. Released daughter radionuclides from 223Ra and 225Ac, for example, can do significant damage to healthy tissue when not retained at the tumour site. Three different approaches to this problem have been proposed: fast uptake and retention of the radiopharmaceutical in tumour cells, local administration, and encapsulation in a nanoparticle.[2] Recent reviews provide overviews of several α-emitters with regards to the challenges posed by recoil and daughter radionuclides, radiation safety concerns, and production and radiochemical separation.[3,4]
This review focusses on the basic radiochemistry of α-emitting radiometals and recent developments in the rapidly growing field of coordination chemistry and ligand design directed towards radiopharmaceuticals for TAT. Tethering the coordination complex to targeting vectors such as monoclonal antibodies (mAbs), fragment antibodies, peptides, and receptor-avid molecules can be used to achieve target tissue selectivity. Recent progress with encapsulating nanoparticles for long-lived multiple α-emitting radionuclides will also be briefly presented. In vivo studies with new α-emitting constructs are briefly discussed to highlight recent progress. Astatine-211 (211At), a radionuclide with potential for TAT, is not covered in the scope of this review because of the markedly different chemistry of astatine compared with radiometals.[5,6]
Actinium
Actinium has three naturally occurring radionuclides. Actinium-227 (227Ac, t1/2 21 years) is the longest-lived isotope of actinium and constitutes almost all naturally occurring actinium. The remainder is made up of trace amounts of actinium-228 (228Ac, t1/2 6.15 h) and 225Ac (kinetic energy of α-particle (Eα) 5.8 MeV), the latter of which is suitable for use with targeting biomolecules with similar biological half-lives, and allows delivery of the radionuclide to clinical locations some distance from the site of production.[7] 225Ac decays to ground-state 209Bi through eight daughter isotopes with a total of four high-energy α-particles emitted (Fig. 1).
Actinium-225 Production
The route of production of 225Ac is primarily through thorium-229 (229Th, t1/2 7920 years), which is sourced from fissile uranium-233 (233U).[8] However, as of 2018, this route has only provided 68 GBq year−1 of 225Ac, and the capacity for scaling up production is limited.[7,9] As a result, alternative methods of producing 225Ac, including accelerator production from thorium-232 (232Th), have been investigated and have shown some promise for increasing availability of 225Ac for clinical use.[10]
Actinium-225 Chemistry
The +3 oxidation state of actinium is the most stable in aqueous solution and therefore the most relevant for biological applications.[11,12] The precise ionic radius of Ac3+ has been historically difficult to determine owing to the scarcity and lack of spectroscopic characterisation. A recent review suggested that the values 1.065 Å (coordination number (CN) 6) and 1.220 Å (CN 9) are the most accurate given the available data.[13] The La3+ ion has similar ionic radii (1.03 Å, CN 6; 1.216 Å, CN 9) and chemical properties to Ac3+, making it a suitable non-radioactive substitute that is easy to handle and is readily available.[12,14] The first hydrolysis constant of Ac3+ has been determined as pKa = 9.4 (La3+ pKa = 9.0), which suggests that neutral and basic buffers are potentially relevant radiolabelling media.[11] Both La3+ and Ac3+ are classified as ‘hard’ acids according to the hard and soft acids and bases (HSAB) theory and therefore prefer non-polarisable, electronegative donor atoms.[15] Complexes of La3+ and Ac3+ range from 3- to 11-coordinate.[16,17] In aqueous solution, larger ions such as La3+ and Ac3+ form 11-coordinate [M(OH2)11]3+ complexes.[17,18] Dissociated or free Ac3+ tends to accumulate in the liver and bone, which can cause serious radiotoxic effects.[19]
Actinium Chelators for Radiopharmaceuticals
A summary of chelators investigated for actinium is presented in Table 1. The widely studied and utilised 2,2′,2′′,2′′′-(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrayl)tetraacetic acid (H4DOTA) macrocycle has historically been the chelator of choice for 225Ac3+ radiopharmaceuticals. The formation constant of the DOTA4– complex with La3+ is 1025.[22] La3+ forms a nine-coordinate complex with the octadentate DOTA4– ligand with the coordination sphere completed by either a bridging O donor atom from an adjacent complex or by an aqua ligand (Fig. 3).[36–38] The coordination geometry of [La(DOTA)(OH2)]– is approximately halfway between capped inverted square prism and capped inverted square antiprism. The average torsion angle between the two planes defined by the four N and four O donor atoms is 22.7°. The solid-state structure is a racemic mixture of the Δ(δ,δ,δ,δ) and Λ(λ,λ,λ,λ) enantiomers.[36]

|

|
Formation of the [225Ac][Ac(DOTA)]– complex required heating to 80°C for 5 min and pH 6 at chelator concentrations >10−6 M to achieve a 99 % radiolabelling efficiency.[14] An efficiency of >95 % has been achieved at milder temperatures (37°C), which are more suitable for conjugation to sensitive biomolecules, but required longer reaction times (2 h) and a higher chelator concentration (>10−3 M).[39] [225Ac][Ac(DOTA)]– is kinetically inert with more than 90 % of the complex remaining intact after 8 days in the presence of a 50-fold excess of La3+ or 7 days in human serum.[14] [225Ac][Ac(DOTA)]– administered intravenously to adult C57BL/6 mice cleared rapidly via the urine, demonstrating that the complex is stable in vivo for at least 5 h.[14,40] Several constructs using [225Ac][Ac(DOTA)]– have subsequently been produced featuring targeting groups, including small molecules, peptides, and antibodies.[21,24–31,39,41–47]
Initial studies to prepare [225Ac][AcDOTA]–-antibody constructs utilised a two-step procedure that involved radiolabelling the bifunctional p-SCN-Bn-H4DOTA (Bn = benzyl) chelator with 225Ac3+ at 55–60°C before attachment to temperature-sensitive antibodies.[48] This process was utilised successfully to prepare the anti-prostate specific membrane antigen (PSMA) construct [225Ac][Ac(DOTA-J591)]–, which was readily internalised by cancer cells and led to tumour regression and prolonged survival of mice bearing prostate carcinoma.[49] This study was integral to the establishment of 225Ac as a applicable radionuclide to deliver α-particles to cancer cells.
Recently, a new two-step method for forming [225Ac][Ac(DOTA–antibody)]– constructs was developed that utilises the bioorthogonal inverse electron-demand Diels–Alder (IEDDA) cycloaddition ‘click’ reaction between a tetrazine (Tz) and trans-cyclooctene (TCO).[31,50] The remarkable selectivity and speed (rate constant k ~1–106 M−1 s−1) of the IEDDA cycloaddition facilitated the efficient addition of polyethyleneglycol (PEG) modified [225Ac][Ac(DOTA-PEG7-Tz)]– to a TCO-modified 5B1 immunoconjugate within 5 min at ambient temperature and pH 6.7 with superior radiochemical yield to the conventional isothiocyanate coupling.[31] The biodistribution of [225Ac][Ac(DOTA-PEG7-5B1)]– in immune-compromised mice bearing BxPC3 (CA19.9 positive, cells expressing carbohydrate antigen 19.9) xenografts demonstrated 32 % injected dose per gram (ID g−1) tumour uptake, and negligible persistent uptake in the kidneys, bone or liver.
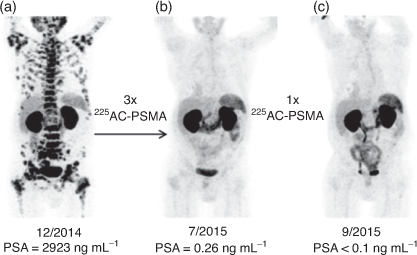
A H4DOTA construct targeting prostate cancer (PSMA-617) has been investigated in clinical trials and demonstrated the extraordinary therapeutic potential of 225Ac3+ to treat widespread tumour growth in vivo. Treatment with 4 × 9–10 MBq (100 kBq kg−1) for one patient resulted in remission (>5 years) from mCRPC (Fig. 4).[30,51] It is important to note that the coordination environment provided by PSMA-617 features the H3DO3A chelator with three acetic acid arms and the fourth used to form an amide bond with the targeting vector. Even subtly different coordination environments can potentially affect the chemical and physical properties of the resulting radiometal complexes.
|
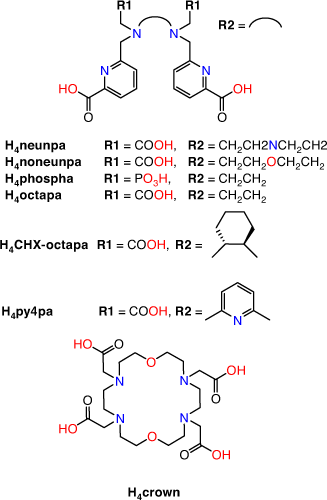
The requisite high temperatures, long reaction times, and/or two-step labelling procedures demonstrate that H4DOTA is not the ideal chelator for use in 225Ac3+ radiopharmaceutical applications. Acyclic chelators have been shown to permit radiolabelling at room temperature; however, the resulting complexes can be more kinetically labile in vivo.[19] Recently, the acyclic picolinate chelators H4neunpa-NH2, H4noneunpa, H6phospa, H4octapa, H4CHX-octapa, and H4py4pa (H4neunpa = 6,6′-(((azanediylbis(ethane-2,1-diyl))bis((carboxymethyl)azanediyl))bis(methylene))dipicolinic acid; H4noneunpa = 6,6′-(((oxybis(ethane-2,1-diyl))bis((carboxymethyl)azanediyl))bis(methylene))dipicolinic acid; H6phospa = 6,6′-((ethane-1,2-diylbis((phosphonomethyl)azanediyl))bis(methylene))dipicolinic acid; H4octapa = 6,6′-((ethane-1,2-diylbis((carboxymethyl)azanediyl))bis(methylene))dipicolinic acid; H4CHX-octapa = 6,6′-((cyclohexane-1,2-diylbis((carboxymethyl)azanediyl))bis(methylene))dipicolinic acid; H4py4pa = 6,6′-(((pyridine-2,6-diylbis(methylene))bis((carboxymethyl)azanediyl))bis(methylene))dipicolinic acid) (Fig. 5) have been investigated for efficient incorporation of 225Ac under mild conditions while also retaining kinetic inertness in vivo. The chelators range from octa- to undecadentate with ether, pyridine, carboxylate, phosphonate or amine donor groups in addition to the bidentate picolinates.
|
The octadentate chelators H4octapa and H4CHX-octapa provide an N4O4 coordination sphere.[21,23] A 10-fold higher ligand concentration was required for H4octapa (10−5 M) to achieve a radiolabelling efficiency of 95 % at ambient temperatures after 1 h compared with H4CHX-octapa (10−6 M). Both 225Ac3+ complexes were stable (>92 % radiochemical purity) in human serum for 7 days at ambient temperatures. The H6phospa chelator had inferior radiolabelling and stability properties to the carboxylate chelators, which may indicate that the lower basicity of phosphonate donors produces weaker ligand–metal interactions with Ac3+.[11,21,48] The in vivo stabilities of these 225Ac3+ complexes are yet to be determined.
The nonadentate chelators H4neunpa-NH2 and H4noneunpa incorporate an additional amine or ether donor in the ligand backbone, respectively. While H4noneunpa achieved >98 % radiolabelling efficiency at ligand concentrations of 10−6 M after 10 min at room temperature, H4neunpa-NH2 was unable to form an 225Ac3+ complex under the same conditions.[20] In the absence of X-ray crystallographic structural data, density functional theory (DFT) calculations of [La(noneunpa)]– suggested a metal ion fully encapsulated by the chelate binding cavity.[20,52] The discrepancies in the radiolabelling performances between H4neunpa-NH2 and H4noneunpa were attributed to the former coordinating in an octadentate manner, thereby allowing the incorporation of an inner-sphere water molecule into the La3+/Ac3+ complex coordination sphere. The ether linkage within the backbone of H4noneunpa allows greater conformational flexibility than the amine linkage of H4neunpa-NH2 to accommodate the large ionic radius of Ac3+. The [225Ac][Ac(noneunpa)]– complex displayed good stability when challenged in human serum at ambient temperature, with a decrease in radiochemical purity of ~10 % over 7 days; however, in vivo stability has yet to be determined.
The undecadentate chelator H4py4pa has a central nitrogen donor atom from a pyridine functional group, two carboxylates, and two picolinate arms, to give an N5O4 coordination sphere. H4py4pa radiolabels with 225Ac3+ to 97 % radiolabelling efficiency at room temperature over 30 min at a chelator concentration of 10−6 M.[22] The [La(py4pa)]– complex remained intact (>99 %) for 9 days in mouse serum and has a formation constant of ~1020.37. DFT calculations for [La(py4pa)]– indicated an 11-coordinate complex encapsulated within the ligand binding cavity with a 2-fold symmetry about the central pyridine. The H4py4pa scaffold was bifunctionalised by incorporating a Bn-NCS group on the central pyridine, which allowed attachment to trastuzumab and in vivo assessment. [225Ac][Ac(py4pa–trastuzumab)]– stayed intact in mouse plasma (97–99 % by instant-TLC (iTLC)) for 11 days at 37°C. Although the immunoreactive fraction of the H4py4pa–trastuzumab bioconjugate decreased to 61 %, the observed tumour uptakes were 23.1 ± 8.8 and 36.9 ± 11.1 % ID g−1 at Days 1 and 6 respectively, followed by a decrease to 17.7 ± 9.3 % ID g−1 at Day 10.
One of the major challenges in the development of Ac3+ radiopharmaceuticals is to provide a scaffold that can better accommodate the large ionic radius of the Ac3+ ion. Bispidines, or 3,7-diazabicyclo[3.3.1]nonanes with acetate, methyl-pyridine or methyl-picolinate pendant groups attached are pre-organised rigid scaffolds that fully encapsulate a metal ion into its preferred coordination geometry. The La3+ and 225Ac3+ complexes were formed with the N6O2 octadentate chelator H2bispa2 (H2bispa2 = 6,6′-({9-hydroxy-1,5-bis(methoxycarbonyl)2,4-di(pyridin-2-yl)-3,7-diazabicyclo[3.3.1]nonane-3,7-diyl}bis(methylene))dipicolinic acid).[23] Radiolabelling with 225Ac3+ proceeds at room temperature and pH 7 to give 94 % radiolabelling efficiency at a concentration of 10−5 M and 64 % at 10−6 M. The La3+ complex has a formation constant of 1011.42. The 225Ac3+ complex remained 89 % intact after 7 days in human serum and 71 % intact after competition with 50 equiv. La3+ for 7 days. In vivo studies employing biological targeting vectors are required to establish the suitability of this chelator for 225Ac3+ targeted therapy.
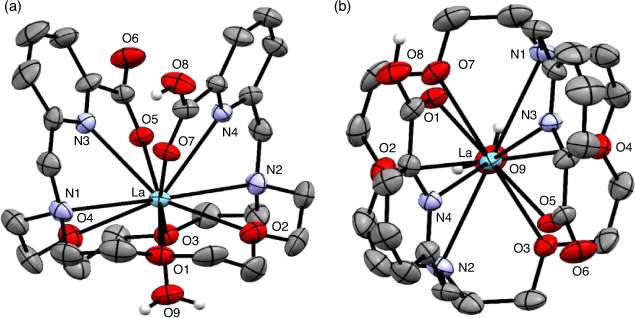
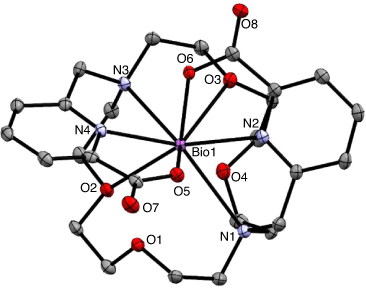
The 1,10-diaza-18-crown-6 macrocycle has been investigated as a suitable scaffold for Ac3+ owing to the cavity size generated by the 18-membered macrocyclic core. The ligand N,N′-bis[(6-carboxy-2-pyridyl)methyl]-4,13-diaza-18-crown-6 (H2macropa) contains two picolinate arms in addition to the two nitrogen and four oxygen donor atoms and forms stable complexes with large lanthanoid ions.[53] X-ray crystallography of the 11-coordinate complex [La(Hmacropa)(OH2)]2+ showed the two pendant arms on one side of the La3+ ion and the N2O4 macrocycle donors and an inner-sphere aqua ligand positioned on the other side (Fig. 6).[14] DFT calculations have shown that protonation of the picolinates increases the coordination of the aqua ligand; however, an aqueous environment decreases the energy gain of H2O coordination and it is therefore unlikely that the 11th coordination site is occupied under biological conditions.[33] The solid-state structure is a racemic mixture of the Δ(δ,λ,δ)(δ,λ,δ) and Λ(λ,δ,λ)(λ,δ,λ) enantiomers.[54] The crystal structures of [La(macrodipa)]+ and [La(Hmacrotripa)]+ both indicate a 10-coordinate complex with the pendant arms folded on either side of the macrocycle and the absence of a coordinating aqua ligand.[55] The formation constants of the [La(macropa)]+, [La(macrodipa)]+, and [La(Hmacrotripa)]+ complexes were determined to be 1014.99, 1012.19, and 1012.57 respectively, indicating the effect of donor atom positioning on the macrocycle.
|
Formation of [225Ac][Ac(macropa)]+ proceeded rapidly at room temperature, resulting in 99 % radiolabelling efficiency after just 5 min at 10−7 M, a concentration a factor of 100 less than in the case with [225Ac][AcDOTA]–.[14,34] Furthermore, Ac3+ remained coordinated by macropa2– for 7 days in the presence of a 50-fold excess of La3+ and analysis of the macropa2– complex in human serum indicated that Ac3+ remained coordinated by macropa2– for 8 days.[14] Biodistribution of [225Ac][Ac(macropa)]+ in mice suggested complex stability with no accumulation of activity in the liver or bone over 5 h.
Bioconjugates of [225Ac][Ac(macropa)]+ were prepared with a small-molecule PSMA-targeting agent by way of an isothiocyanate functional group. Radiolabelling of the macropa–PSMA construct with 225Ac3+ proceeded at ambient temperatures and pH 5 with 98 % radiolabelling efficiency after 20 min. Biodistribution in LNCaP (prostate cancer) tumour xenograft-bearing mice demonstrated that initial uptake in the kidneys and tumour (52 ± 16 and 12.8 ± 3.1 % ID g−1, respectively) after 4 h was followed by significant kidney clearance (1.5 ± 0.1 % ID g−1) and some tumour retention (4.9 ± 0.5 % ID g−1) at 96 h. Importantly, no accumulation in the liver, spleen, and bone was observed over several days.
The 4,7,13,16-tetra-aza-18-crown-6 derivatised with four acetic acid groups, 2,2′,2′′,2′′′-1,10-dioxa-4,7,13,16-tetraazacyclooctadecane-4,7,13,16-tetrayl-tetraacetic acid, and given the trivial name ‘H4crown’ (Fig. 5), contains a possible 10-coordinate N4O6 coordination sphere; however, the nature of La3+ or Ac3+ coordination is still unknown.[35] Radiolabelling of H4crown with 225Ac3+ proceeded at ambient temperature between pH 5 and 7 over 10 min with 96 % radiolabelling efficiency at concentrations down to 10−6 M but with negligible labelling at 10−7 M. The chelator was tethered to an α-melanocyte-stimulating hormone (αMSH) targeting agent at one of the carboxylate pendant arms. This method of bioconjugation has the potential to alter the coordination environment and properties of the resulting complex. However, radiolabelling of the bioconjugate with 225Ac3+ proceeded under the same conditions as H4crown, and the complex remained approximately 90 % intact in human serum after 8 days at 37°C. The biodistribution of [225Ac][Ac((crown)–αMSH)] prepared 4 or 18 h before injection was evaluated in mice bearing B16F10 melanoma tumours 2 h post injection.[35] The construct prepared 18 h before injection demonstrated significantly lower tumour uptake, which was attributed to radiolytic degradation of the bioconjugate. Interestingly, radioTLC did not indicate radiolysis, thus demonstrating the need to use additional quality control methods such as radioHPLC to determine the integrity of 225Ac3+ radiopharmaceuticals. Most notably, the results indicated the need for immediate injection after preparation of the radiopharmaceutical, which raises concerns about the feasibility of using centralised radiopharmaceutical synthesis with distribution to clinical locations.[49]
A summary comparison of 225Ac3+ activity incorporation v. chelator concentration is given in Fig. 7. The recent developments in Ac3+ chelating scaffolds have brought about a significant improvement in the radiolabelling efficiencies now possible compared with H4DOTA. The poorly understood coordination chemistry of Ac3+ has benefited from the utilisation of X-ray crystallography with La3+ and DFT calculations. A recent investigation utilising spectroscopic characterisation (extended X-ray absorption fine structure (EXAFS) and NMR) of the longer-lived 227Ac3+ complexes suggested that these techniques will become important tools for investigating Ac3+ coordination chemistry and advancing Ac3+ chelator development.[24,56]
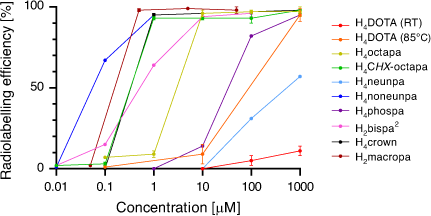
|
The most successful new ligands to date, such as H2macropa and H4py4pa, have been bifunctionalised and incorporated into constructs that have shown significant promise for targeting tumours with excellent in vivo stability. Further development and validation will require pre-clinical and clinical studies focusing on the anti-tumour efficacy of these chelators as 225Ac-based radiopharmaceuticals.
Bismuth
There are two radionuclides of bismuth with potential applications in α-therapy: 213Bi (Eα(213Po) 8.4 MeV, gamma (γ) 435 keV) and 212Bi (Eα 8.8 and 6.1 MeV).[58,59] The particle energy released per disintegration of 213Bi comprises 92.7 % from α and 7.3 % from beta-electron (β–) emission. Both 212Bi and 213Bi deliver high radiation doses within a short timeframe and have been investigated with large slower-circulating antibodies as well as small bioconjugates with rapid blood clearance.[60–62] Significant challenges with supply, high cost, and radioprotection issues are notable and have been discussed in recent comprehensive reviews.[9,63,64] The 440-keV photon γ-ray emitted from 213Bi has been used for preclinical and clinical single-photon emission computed tomography (SPECT).[65,66] The cyclotron-produced radionuclides bismuth-205 (205Bi, t1/2 15.3 days) and bismuth-206 (206Bi, t1/2 6.2 days) have been used as longer-lived surrogates to investigate the radiochemistry of bismuth chelators.[67]
Bismuth-212/213 Production
212Bi is generated by way of the natural thorium (232Th) decay chain and is the direct daughter radionuclide of 212Pb. All clinical studies with 213Bi to date have used 225Ac sourced from 229Th.[68,69] However, the investigation of accelerator-based production routes has the potential to increase the reliable production and delivery of 225Ac for both 225Ac and 213Bi radiopharmaceuticals.[9] Clinical availability of 213Bi is through an 225Ac/213Bi generator whereby 225Ac is retained on a cation exchange resin and 213Bi is eluted as [213Bi]BiI4– and [213Bi]BiI52– using a mixture of 0.1 M HCl/0.1 M NaI. The generator can be distributed and stored for weeks providing in-house 213Bi production.[7,70]
Bismuth Chemistry
Aqueous bismuth chemistry is dominated by the trivalent oxidation state, with the resulting 6s2 valence configuration producing examples of the inert pair effect in some coordination complexes.[71] At low pH (beginning at pH 0), Bi3+ readily undergoes hydrolysis to form Bi3+ hydroxides.[72] Interestingly, radiolabelling with 213Bi3+ can be performed at pH values ranging from 4.0 to 10 owing to the formation of BiI4–/BiI52– after elution with 0.1 M HCl/0.1 M NaI.[73] Trivalent bismuth is a borderline Lewis acid according to HSAB theory.[15] The Bi3+ ion exhibits a variety of coordination numbers and irregular coordination geometries with oxygen and nitrogen donor atoms as well as thiolate groups.[74,75] Coordination numbers over the range 3–10 have been reported for Bi3+ complexes with the typical octadentate ionic radius reported as 1.17 Å.[72,76] Dissociated or free radioactive Bi3+ tends to accumulate predominantly in kidney with slow clearance kinetics.[77–79] Bi3+ is known to bind to Zn2+ sites (e.g. metallothionein) and Fe3+ sites (e.g. transferrin) in proteins.[80]
Bismuth Chelators for Radiopharmaceuticals
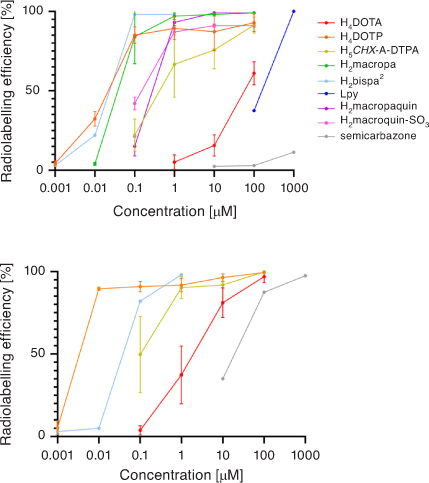
A summary of chelators investigated for bismuth is presented in Table 2. The short half-lives of 212Bi and 213Bi require fast radiolabelling (ideally <5 min) and quantitative yields to facilitate good manufacturing practice (GMP) production. Whether this can be achieved at ambient or elevated temperatures depends on the chelator and the robustness of the targeting agent. Early studies with bismuth radionuclides used bifunctional derivatives of diethylenetriaminepentaacetic acid (H5DTPA) to radiolabel antibodies and were instrumental in demonstrating the potential of α-emitting radionuclides attached to mAbs. Despite the fact that bifunctional H5DTPA–mAb constructs were radiolabelled under mild conditions and [Bi(DTPA)]2– was extremely thermodynamically stable (formation constant 1035.6), the chelators were shown to be insufficiently inert in vivo for use in targeting α-therapy.[79,93] The introduction of a trans-cyclohexyl unit increased steric rigidity and provided a pre-organised geometry of donor atoms that improved in vivo complex inertness while retaining rapid formation kinetics.[94] The advantages of the bifunctional derivative [Bi(p-SCN-Bn-CHX-A-DTPA)]2– (formation constant 1034.9–1035.6) were demonstrated in a study that conjugated the chelator to an immunoglobulin IgG mAb HuM195 (anti-CD33).[83] The H5CHX-A-DTPA-HuM195 construct showed moderately efficient radiolabelling with 213Bi (78 % ± 10 %) in only 10 min at ambient temperature. The [205/206Bi][Bi(CHX-A-DTPA-HuM195)] construct showed reduced uptake in kidney compared with both free bismuth and the analogous H5DTPA construct.

|
Efforts to develop bismuth chelators with greater kinetic stability naturally also focussed on macrocyclic polyaminocarboxylic acids such as H4DOTA. The crystal structure of the eight-coordinate [Bi(DOTA)]– complex has an average torsion angle between the squares determined by the four O and four N donor atoms of the ligand of 25.9°, which indicates a twist from the ideal square antiprismatic arrangement of 45° (Fig. 8). The solid-state structure is a racemic mixture of the Δ(δ,δ,δ,δ) and Λ(λ,λ,λ,λ) enantiomers.[86]

|
[Bi(DOTA)]– has a formation constant of 1030.3 and is sufficiently inert to provide a few hours of stable coordination commensurate with the short radionuclide half-life.[96] Monoclonal antibodies incorporating [205/206Bi][Bi(DOTA)]– were shown to be more stable in vivo than the [205/206Bi][Bi(DTPA)]2– constructs by reduced kidney uptake and increased tumour uptake.[97] However, H4DOTA demonstrated slow radiolabelling kinetics and low radiolabelling efficiency under conditions suitable for sensitive targeting molecules (61 % radiolabelling efficiency, room temperature (RT), pH 5.5, 5 min). Radiolabelling efficiency of >96 % was achieved with heating to 95°C at chelator concentrations >10−4 M (Table 2).[98] Although H4DOTA is not conducive to rapid Bi3+ radiolabelling of heat-sensitive antibodies, the safety, feasibility, and effectiveness of 213Bi therapy have been demonstrated with targeting peptides and molecules for treating neuroendocrine and prostate tumours (Fig. 9).[99–101] Furthermore, treatment has been more effective than β-emitting radionuclide therapy in some cases.[62,102] Typical 213Bi3+ radiolabelling conditions with H4DOTA-peptide constructs such as DOTA-tyrosine-3-octreotate (DOTA-TATE) consist of high temperatures (95°C for 5 min, often with a microwave synthesiser) at pH 8.7 and concentrations ~10−5 M to achieve quantitative yields with radiochemical purity ≥85 %.[100] The whole labelling procedure takes on average 20 min to produce a ready-to-inject radiopharmaceutical in a vial.[73]
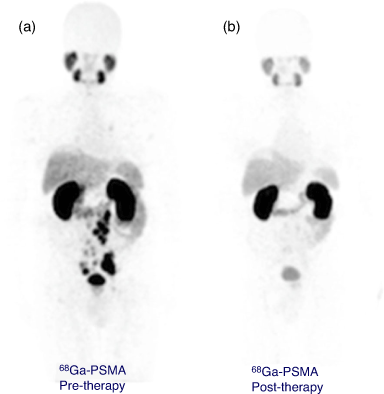
|
The phosphonic acid macrocycle H8DOTP forms an eight-coordinate twisted square antiprism with Bi3+ (Fig. 8).[95] The [Bi(H4DOTP)]– complex is highly symmetrical, with C4 symmetry and an average twist angle of 25.4°. The solid-state structure was a racemic mixture of the Δ(δ,δ,δ,δ) and Λ(λ,λ,λ,λ) enantiomers. NMR spectroscopy indicated that the complex interconverts between the two enantiomeric pairs in solution.[89] The [Bi(DOTP)]5– complex was shown to have a formation constant of 1038.7 and significant kinetic inertness (t1/2,pH 3 = 47000 h).[89] H8DOTP displayed an impressive ability to achieve 213Bi radiolabelling efficiencies of ~90 % at RT and ligand concentrations of 10−4 M that was comparable with CHX-A-DTPA under the same conditions.[87] Comparison stability studies showed that [213Bi][Bi(H4DOTP)]– was more stable than [213Bi][Bi(DOTA)]– and [213Bi][Bi(CHX-A-DTPA)]2– in both human plasma and to an excess of DTPA5– over 3 h. The faster formation kinetics could be due to the affinity of the Bi3+ ion for phosphonate oxygen atoms and/or the orientation of the phosphonate groups (two above and two away from the ring N atoms), which is in contrast to H4DOTA, where all four carboxylates are positioned above the plane of the ring N atoms.[95,103] The results would suggest bifunctional chelators based on H8DOTP should be investigated to radiolabel antibodies with 213Bi and compared in vivo with H5CHX-A-DTPA and H4DOTA. A cyclen derivative bearing four pyridine groups (Lpy) showed higher selectivity for Bi3+ over Ac3+ compared with H4DOTA and H5CHX-A-DTPA, which could be useful to minimise detrimental dose effects of small quantities of parent 225Ac from a generator.[90]
2-(4,7-Biscarboxymethyl[1,4,7]triazacyclonona-1-yl-ethyl)carbonyl-methylamino]acetic acid (NETA) and {7-[2-(bis-carboxymethyl-amino)ethyl]-4,10-bis-carboxymethyl-1,4,7,10-tetraaza-cyclododec-1yl}-acetic acid (DEPA) possess both acyclic and macrocyclic frameworks to promote rapid formation kinetics at ambient temperatures with high thermodynamic and kinetic stability (Fig. 10). Radiolabelling investigations with both chelators have thus far been limited to the use of 205/206Bi with trastuzumab radioimmunoconjugates achieving >93 % incorporation after 1 min at pH 5.5 and RT.[61,104] The 205/206Bi radioimmunoconjugates are very stable in human serum (up to 7 days) and showed high in vivo stability in athymic mice bearing LS174T xenografts with tumour to kidney ratios of 4–5 24 h post injection. NETA and DEPA are potential octadentate and decadentate chelators, respectively; however, structural characterisations of the bismuth complexes are yet to be reported. The encouraging in vivo results demonstrate that they remain promising chelators for 213Bi TAT.
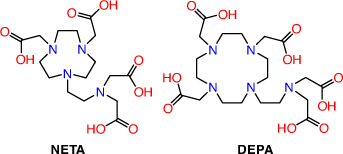
|
Recently, the Bi3+ complexes of dimethyl-cyclen derivatives bearing two bidentate picolinic acid groups (H2Me-do2pa) or two bidentate semicarbazone groups have been reported. The crystal structure of [Bi(Me-do2pa)]+ revealed C2 symmetry and a twisted-square antiprismatic coordination geometry with an average twist angle of 21.2°.[91] The complex [Bi(semicarbazone)]3+ has an average twist angle of 12.8° and a less ordered and symmetrical structure compared with the previously mentioned Bi3+ cyclen derivatives.[92] The 213Bi complexes of the dimethyl-cyclen derivatives were produced in 5 min at 90–95°C and were stable in serum for at least two decay half-lives. Both Bi3+ complexes also were reported to have increased stability over H4DOTA towards acid dissociation.[92,105]
Bismuth complexes of diaza-18-crown-6 ethers bearing picolinates, phosphonates, and quinolinols were characterised by X-ray crystallography and revealed distorted pentagonal pyramidal coordination geometries arising from the stereochemically active 6s2 lone pair (Fig. 11).[85] The kinetic inertness of the complexes increased as a function of donor atom basicity and increasing covalency of the Bi–donor atom bonds. A compromise between faster, more efficient radiolabelling over kinetic inertness would require consideration of both the short half-life of 213Bi and the pharmacokinetics of the resulting radiopharmaceutical.
|
Heptadentate and octadentate (bispa2) bispidine chelators featuring acetate pyridine and picolinate pendent groups form nine-coordinate Bi3+ complexes with the coordination spheres completed by a coordinating nitrate anion.[84] Radiolabelling efficiency with 213Bi was superior to H5CHX-A-DTPA and H4DOTA at lower concentrations at both 95° and 25°C. The heptadentate ligand produced 213Bi complexes that were slightly less inert than H4DOTA to a transchelation challenge with an excess of DTPA5–. The potential of nonadentate ligands that complete the coordination sphere may increase the kinetic inertness of the 213Bi complexes.
A summary comparison of 213Bi3+ radiolabelling efficiency versus chelator concentration is given in Fig. 12. Acyclic and macrocyclic chelators have been investigated over recent years for use with Bi3+ radionuclides, with several showing impressive radiolabelling efficiency and kinetic inertness in human plasma. 212/213Bi3+ may also benefit from chelators that incorporate a range of hard, borderline, and soft Lewis base donor atoms. The limited availability, short half-lives of 212/213Bi3+, and α-emitting daughter radionuclides remain a challenge. Further work is also needed to develop bioconjugates incorporating chelators other than H4DOTA.
|
Lead
212Pb is a β– emitting radionuclide and is the direct parent isotope of 212Bi.[106] The use of 212Pb effectively prolongs the half-life of 212Bi in vivo, more closely matching the pharmacokinetics of various targeting biomolecules.[107] In theory, the decay of 212Pb should not result in the release of 212Bi because the calculated recoil energy of 0.5 eV from the β– decay is insufficient energy to cause the breaking of bonds (10 eV).[108] However, the internal conversion of ~30 % of γ-rays emitted by the decay of 212Pb results in highly ionised products sufficient to cause radionuclide demetallation from the chelator.[88] As a result, 212Bi released from the decay of 212Pb often accumulates in the kidneys. 212Pb has the advantage that the same element can be used to perform dosimetry calculations using the radionuclide 203Pb, a γ-emitting isotope (279 keV; 81 %) with a half-life of 51.9 h suitable for SPECT imaging.[58]
Lead-212 Production
224Ra-based generators allow the elution of either 212Bi or 212Pb (depending on eluent composition).[63,106] 224Ra is isolated from the decay chain of 232U and the daughter isotope thorium-228 (228Th, t1/2 1.9 years), which means it can be obtained on an industrial scale over many years.[109] 228Th produced from a 500 MeV cyclotron has been used to build a 228Th/212Pb generator with the ability to produce up to 10 MBq of 212Pb daily.[110] Production of 203Pb occurs through proton irradiation of naturally occurring thallium.[111]
Lead Chemistry
Lead is primarily found in the +2-oxidation state in aqueous environments. Hydrolysis is not an issue under acidic conditions (pKa 7.7); however, Pb2+ forms the mononuclear hydrous oxides [Pb(OH)]+, [Pb(OH)2], and [Pb(OH)3]– in dilute aqueous solutions in the pH range 6–13.[112] The effective ionic radius of Pb2+ in eight-coordinate complexes is 1.29 Å.[12] Pb2+ is classified as a borderline Lewis acid by the HSAB theory, and exhibits irregular coordination geometries ranging from 1- to 12-coordinate with oxygen, nitrogen, and thiolate groups.[15,113]
Lead Chelators for Radiopharmaceuticals
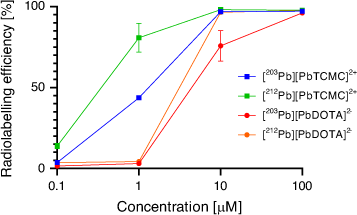
A summary of chelators investigated for lead is presented in Table 3. Chelators investigated for 212Pb/212Bi in vivo generators have included H4DOTA and the tetraacetamide cyclen derivative DOTAM aka TCMC (1,4,7,10-tetraaza-1,4,7,10-tetra(2-carbamoyl methyl)cyclododecane). H4DOTA forms an eight-coordinate complex with Pb2+ with an N4O4 coordination sphere (Fig. 13).[115] The metal ion in the complex is situated closer to the N4 plane than the O4 plane by 0.677 Å and the complex has a distorted geometry with a torsion angle of 22.5° between the two planes. The solid-state structure is a racemic mixture of the Δ(δ,δ,δ,δ) and Λ(λ,λ,λ,λ) enantiomers. The [Pb(DOTA)]2– complex has a formation constant of 1024.3.[125]

|

|
A bioconjugate with an α-MSH targeting moiety, H4DOTA-Re(Arg11)CCMSH, was radiolabelled with 212Pb.[127,128] Although 30 % of 212Bi was lost from the chelator, analysis of the biodistribution of [212Pb][Pb(DOTA-Re(Arg11)CCMSH)]2– indicated that over the course of 24 h non-specific uptake of 212Pb or 212Bi was not observed. This suggests that once localised, 212Bi lost from the chelator remains localised within the cell, which demonstrates the importance of rapid blood clearance and tumour internalisation.
Two crystal structures of the Pb2+ complex of DOTAM have been published. The structures both showed the eight donor atoms encapsulating the ion but differed by the presence or absence of a water molecule weakly interacting with the Pb2+ ion (Pb–O = 3.52 Å).[126,129] Whether the presence of the water molecule was indicative of an interaction with a stereochemically active lone pair has been the subject of some debate because of the effect it might have on the thermodynamics of complex formation.[115] The resulting chiral complex has an average torsion angle of 26° (Fig. 13).[126] The solid-state structure is a racemic mixture of the Δ(δ,δ,δ,δ) and Λ(λ,λ,λ,λ) enantiomers. An accurate formation constant has not been determined; however, a lower-limit formation constant of 1019 has been calculated.[130]
The [Pb(DOTAM)]2+ complex has been shown to be more inert to demetallation under acidic conditions (pH 3.5) than [Pb(DOTA)]2–.[117] Radiolabelling of the bifunctional DOTAM derivative p-SCN-Bn-TCMC (Fig. 14) with 203Pb resulted in >95 % radiolabelling efficiency at 37°C in 30 min at pH 5–6 and 10−4 M.[116]
|
The [203Pb][Pb(p-SCN-Bn-TCMC)] complex remained 83 % intact in serum at 37°C for 2 days. The chelator p-SCN-Bn-TCMC demonstrated superior incorporation of both 212Pb and 203Pb compared with H4DOTA at pH 7 and room temperature after 1 h (Fig. 15). Interestingly, the DOTAM mechanism of Pb2+ coordination was proposed to consist of an arm-by-arm chelation process leading to successive five-membered ring closure as opposed to the fast formation of a PbO4 intermediate in which all four carbonyl groups are bound as in the case of H4DOTA.[126]
|
Radiolabelling of p-SCN-Bn-TCMC-trastuzumab with 212Pb from an 224Ra/212Pb generator achieved a >95 % radiolabelling efficiency at pH 5–6 and 37°C for 1 h without stirring, or 30 min at 750 rpm at concentrations between 0.15 and 1 mg L−1.[122,131] Approximately 70 % of the 212Bi remained associated with the complex after 212Pb decay. Simultaneous radiolabelling in a solution with the daughter isotopes resulted in 70 % radiolabelling efficiency with 212Bi over 5 min, which increased to 86 % after 30 min, indicating slower reaction kinetics with 212Bi compared with 212Pb.[122] The first in-human trial of [212Pb][Pb(p-SCN-Bn-TCMC-trastuzumab)]2+ demonstrated tolerable toxicity and tumour regression at a dosage of 21.09 MBq m−2.[121]
PSMA is an attractive targeting molecule for 212Pb because the mechanism of PSMA targeting involves internalisation into the cell, which would minimise the translocation of released 212Bi due to 212Pb decay.[108] Several PSMA-targeting conjugates have been reported that utilise the chelators H4DOTA, p-SCN-Bn-TCMC or the triacetamide acetic acid H3DO3AM for radiolabelling with 203Pb and/or 212Pb. p-SCN-Bn-TCMC-PSMA derivatives have been radiolabelled using solutions of 212Pb from an 224Ra/212Pb generator or in an 224Ra-solution in transient equilibrium with daughter nuclides.[108,119,120] The complexes are stable, remaining intact for 72 h in human serum at 37°C.[116] DOTA4–-based conjugates tended to display higher renal retention than the positively charged agents that use TCMC as the chelator.[119] In an athymic mouse model bearing human prostate C4–2 xenografts, a DOTA4–-based conjugate demonstrated 18 % ID g−1 in tumour tissue versus 53 % ID g−1 in the kidneys, while the p-SCN-Bn-TCMC-based conjugate achieved similar tumour uptake with tumour to kidney ratios 2.5 times higher.[120] A SPECT study in two patients using 203Pb-labelled PSMA ligands demonstrated the potential for TAT of prostate cancer with 212Pb but also cautioned towards the inherent limitations of using imaging for dosimetry estimates owing to the release of daughter nuclides.[108,116]
New chelators have been proposed for radiolabelling with Pb2+ radionuclides such as pyridine-based cyclen derivatives that exhibited the ability to complex 212Pb/203Pb at ambient temperature at pH 7.[110] The coordination chemistry of the Pb2+ complexes have not yet been reported. The Pb2+ coordination chemistry of Me-dod2pa and the bis(semicarbazone) macrocyclic chelator have been investigated in detail.[105,132] Both chelators form eight-coordinate complexes with Pb2+ with an N6O2 coordination sphere. The Pb2+ lone pair in both complexes appears to be stereochemically inactive. Formation of the non-radioactive [Pb(Me-dod2pa)] and [Pb(semicarbazone)]2+ complexes proceeds quickly at pH 7.4 with half-lives of 13.8 and 20.0 min under pseudo-first-order conditions, respectively. Both Pb2+ complexes demonstrated resistance to transchelation in the presence of acid and a 100-fold excess of ethylenediaminetetraacetic acid (EDTA) and are promising ligands for radiolabelling studies.
An octadentate [2.2.2]-cryptand (Fig. 14) and one bidentate perchlorate ion formed an N2O6 10-coordinate complex with Pb2+.[124] 1H NMR of the Pb2+ complex indicated the presence of a single, highly symmetrical isomer in solution. The [2.2.2]-cryptand was radiolabelled with 203Pb (pH 7, RT, 60 min) with efficiencies of >99 % and 88.6 % ± 6.0 % at chelator concentrations of 10−5 and 10−6 M, respectively. A bifunctional [2.2.2]-cryptand derivative was conjugated to trastuzumab and radiolabelled with 203Pb and demonstrated in vitro stability in human serum over 72 h (97.1 % ± 0.56 % intact). In vivo experiments would validate bifunctional [2.2.2]-cryptands as promising chelators for Pb radiopharmaceuticals.
Amide derivatisation of H5DTPA generated the pentaacetamide DTPAm, which formed an eight-coordinate complex with Pb2+ with a formation constant of 108.79.[114] X-ray diffraction studies of [Pb(DTPAm]2+ showed that the lone pair was active and a hemidirected complex was formed. Radiolabelling with 203Pb proceeds rapidly, giving a 95 % radiolabelling efficiency at RT in 15 min at pH 7 with a chelator concentration of 10−5 M. In vitro and in vivo stabilities have not been assessed; however, the vacant coordination site is expected to promote transmetallation of Pb2+ to biomolecules with suitable donor atoms.
The use of a generator allows production of 212Pb over many years. The Pb2+ DOTAM/p-SCN-Bn-TCMC coordination chemistry is well understood, and the chelators have the ability to radiolabel sensitive biomolecules and small molecules with 203Pb and 212Pb that have been validated in pre-clinical and clinical studies. The loss of 212Bi on 212Pb decay remains a significant issue, with kidney being the dose-limiting organ. Radionuclide release also affects prospective dosimetry calculations using SPECT for the matched 203/212Pb theranostic pair. Attempts have been made to use acyclic and macrocyclic chelators to ‘re-capture’ released 212Bi but have thus far demonstrated limited success owing to the concentrations required to compete with ions present in blood serum.[133,134]
Terbium
149Tb (Eα 4.0 MeV) emits both positrons (β+) and α-particles, which allows for simultaneous α-therapy and positron emission tomography (PET), or α-PET. Notably, 149Tb is the only α-emitting radiometal discussed without α-emitting daughter isotopes, which reduces the potential for non-target exposure to harmful ionising radiation. Although the precise biodistribution of free terbium within the body has not been determined, uptake of free radio-lanthanoids in vivo has been observed in the bone and liver.[135]
Terbium-149 Production
149Tb can be produced through proton-induced spallation of tantalum, but difficulties with purification have led to production through the irradiation of early lanthanoid targets, such as the 152Gd(p,4n)149Tb reaction.[136–138] The most significant issue with the use of 149Tb3+ in radiopharmaceuticals remains availability owing to the specialist facilities required for production, which has hindered efforts to study the radiochemistry.[135] However, as Tb3+ radionuclides continue to show promise for radiopharmaceutical applications, the construction of more facilities may allow availability of the radionuclides on a large scale.
Terbium Chemistry
Terbium exists primarily in the +3 oxidation state, although it has been recently isolated in both the +2 and +4 oxidation states.[139–141] Owing to lanthanoid contraction, the ionic radius of Tb3+ (1.040 Å, CN 8) is significantly smaller than La3+ (1.160 Å, CN 8).[12] Tb3+ forms eight-coordinate complexes in aqueous media.[142] Hydrolysis of Tb3+ is generally insignificant under radiolabelling conditions owing to the high pH at which hydroxide formation occurs (pKa = 7.9).[143] Tb3+ is defined as a hard acid by the HSAB theory and has a varied coordination chemistry, forming complexes with 1–12 donor atoms with a preference for a coordination number of 8–9.[144] Terbium exhibits a preference for oxygen donor atoms, characteristic of the hard acid lanthanoid metal centre. According to a search of the Cambridge Crystallographic Data Centre (CCDC) database in 2021, 94 % of terbium-containing crystal structures and 87 % of all lanthanoid-containing crystal structures were reported to have at least one oxygen donor atom.
Terbium Chelators and Radiopharmaceuticals
A summary of chelators investigated for terbium is given in Table 4. A rituximab antibody conjugated with H5CHX-A-DTPA was radiolabelled with 149Tb at RT, pH 5.5, over 10 min at concentrations >10−3 M to give >99 % radiolabelling efficiency.[135] The α-emitting bioconjugate prevented tumour growth in 89 % of mice for 120 days after intravenous graft with Daudi cells. The suitability of H4DOTA as a chelator for Tb3+ has been reviewed.[143,149] H4DOTA forms a complex with Tb3+ with a formation constant in the region of 1023.6–1027.0.[143] The solid-state structure of the complex has not been reported to date. The solid-state crystal structure of [Tb(HDO3AP)]– (H5DO3AP, 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic-10-methylphosphonic acid) has provided some insights into the coordination chemistry of cyclen-based Tb3+ complexes (Fig. 16).[150] The terbium metal centre bonds with the four nitrogen and four oxygen donor atoms of the chelator and a nearby water molecule to give a nine-coordinate N4O5 coordination sphere. The Tb–O bond lengths are shorter than the Tb–N bond lengths by an average of 0.32 Å. The average twist between the two planes constructed from the two sets of macrocyclic donor atoms is 27°, which gives a monocapped twisted square anti-prism coordination geometry. The solid-state structure is a racemic mixture of the Δ(δ,δ,δ,δ) and Λ(λ,λ,λ,λ) enantiomers.

|

|
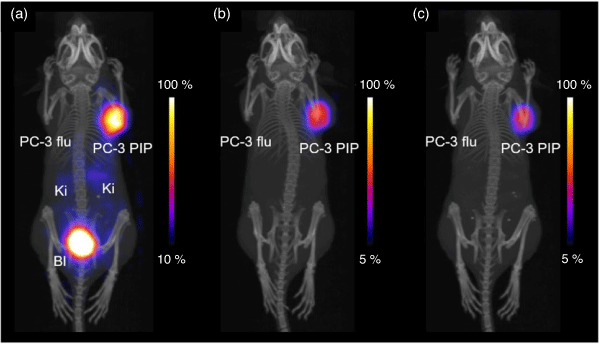
Radiolabelling of the H3DO3A-based chelator PSMA-617 with 149Tb (95°C, 15 min, pH 4.5, 10−3 M) achieved radiochemical purity >98 %.[147] Treatment with 6 MBq of [149Tb][Tb(PSMA-617)] significantly reduced tumour growth by 82–87 % in mice bearing PC-3 PIP tumours and increased the median lifetime from 20 days in untreated mice to 36 days when injected on two consecutive days. PET imaging with the same complex confirmed the selective accumulation of the isotope in tumour xenografts (Fig. 17). These results support the potential use of 149Tb isotope in simultaneous α-PET.
|
Preliminary investigation of the Tb3+ complexes of H2macropa, H2macrodipa, H3macrotripa, and H2CHX-macropa showed the complexes have formation constants of 1011.79, 109.68, 1010.19, and 1010.98, respectively.[151] The ionic radius of Tb3+ suggests it is likely to form an eight-coordinate complex, a conclusion supported by DFT calculations.[12,55] It remains to be determined whether mild radiolabelling conditions and kinetic inertness can be achieved with these chelators.
Accessibility remains a barrier to the development of 149Tb radiopharmaceuticals. Advances in Tb3+ coordination chemistry and radiochemistry will hopefully help promote this promising radionuclide.
Thorium
227Th (Eα 6.0 MeV) emits five high-energy α- and two β–-particles in its decay pathway.[152] The additional co-emission of γ-rays may also allow simultaneous SPECT imaging.[153] The most significant advantage of 227Th compared with other α-emitting isotopes is the availability of the isotope on an industrial scale. However, the emission of five α-particles and two β–-particles by daughter isotopes along the decay pathway complicates the use of 227Th as a therapeutic isotope owing to the potential for off-target side effects.
Thorium-227 Production
The parent isotope 227Ac is already used to access 223Ra. The long half-life of 227Ac allows the development of an 227Ac/227Th generator.
Thorium Chemistry
Thorium is found almost exclusively in the +4-oxidation state.[139,154] Th4+ has an ionic radius of 0.94 Å in six-coordinate complexes and 1.05 Å in eight-coordinate complexes.[12] Thorium has been observed in coordination complexes with CNs ranging from 5 to 12.[155] In aqueous solutions, Th4+ forms 8–10-coordinate complexes with aqua ions at pH < 3 and remains the predominant form between pH 5 and 8, but forms hydroxides at pH ≥ 4.[156,157] As an actinoid, Th4+ is classified as a hard acid by the HSAB theory.[16] Free 227Th4+ has been shown to accumulate in bone.[158,159]
Thorium Chelators and Radiopharmaceuticals
A summary of chelators investigated for thorium is given in Table 5. Both acyclic and macrocyclic chelators have been investigated for 227Th radiopharmaceuticals, with the majority exclusively employing oxygen donor atoms apart from H4DOTA (Fig. 18). Thorium formed a nine-coordinate complex with DOTA4– and one solvent dimethylsulfoxide (dmso) molecule (Fig. 19).[167] The synthesis of [Th(DOTA)(dmso)] was found to only proceed in water-free environments. The resulting complex was a capped square antiprism, with an average torsion angle of 38° between the N4 and O4 planes. The Th4+ metal centre is significantly closer to the O4 plane (0.542 Å) than the N4 plane (1.770 Å). The solid-state structure is a racemic mixture of the Δ(δ,δ,δ,δ) and Λ(λ,λ,λ,λ) enantiomers.

|

|

|
The [Th(DOTA)] formation constant has not been determined. A competition experiment between a Th-Arsenazo III complex and DOTA4– to determine the formation kinetics did not proceed at room temperature for more than 2 months, but proceeded if heated to 100°C.[162] The slow formation kinetics were confirmed when radiolabelling of a p-SCN-Bn-DOTA4– mAb construct with 227Th required incubation overnight at 37°C in pH 5 buffered aqueous solution to give 99 % radiolabelling efficiency.[168] The concentration of the conjugate was unknown owing to the variable chelator-to-bioconjugate ratio.
An octadentate chelator consisting of a combination of macrocyclic and acyclic terephthalamide groups forms an eight-coordinate 232Th4+ complex with a distorted dodecahedron geometry (Fig. 20).[162] The complex has a very high formation constant of 1054 and the formation kinetics (t1/2 57 s) are rapid. To date, the 227Th radiolabelling conditions and stability of the 227/232Th complex under biological conditions have not been reported.

|
The majority of chelators that have been investigated for use with 227Th are acyclic chelators with bidentate hydroxypyridinone (HOPO) or catecholamide (CAM) groups. These chelators are generally composed of a polyamine scaffold to which the oxygen atom donor groups are attached. The octadentate chelator H4(3,4,3-LI(1,2-HOPO)) consists of four 1-hydroxy-2-pyridinone groups. The Th4+ complex has a formation constant of 1040.1 and was shown to be kinetically inert to transmetallation against DTPA ([Th(DTPA)]– formation constant 1028.7).[161] The optimised structure of [Th(3,4,3-LI(1,2-HOPO)] was calculated using DFT with the eight Th4+–O distances consistent with the experimentally determined distances of [Th(1,2-HOPO)4].[169,170] A chelator with the same polyamine scaffold but featuring four N-methyl-3-hydroxypyridine-2-one functional groups demonstrated good radiolabelling capability, achieving 70 % radiolabelling efficiency over 10 min at pH 5.5, which increased to 83 % after 2.5 h.[160] The resulting complex was stable in phosphate-buffered saline (PBS) at ambient temperature, 95 % remaining intact after 6 days, and 73 % after 14 days.
The octadentate bifunctional chelator H4(Me-3,2-HOPO-OH) consists of four N-methyl-3-hydroxypyridine-2-one functional groups and a carboxylic acid for bioconjugation.[163] The [Th(Me-3,2-HOPO-OH)] complex has a formation constant of 1041.7 and displays charge-dependent selectivity for 227Th4+ over other actinoids and biologically relevant metals of other oxidation states. Radiolabelling of H4(Me-3,2-HOPO-OH) with 227Th proceeds over 30–60 min at RT. These mild conditions allow bifunctional derivatives to be prepared with both antibody and small peptide bioconjugates.[161,164,171,172] The kinetic stability of Th4+ complexes can be indirectly determined by monitoring uptake in bone owing to the known biodistribution of free Th4+.[158] The biodistribution of [Th(Me-3,2-HOPO-OH)] in female C57B16 mice indicated minimal uptake in the bone (0.33 % ID g−1 in the femur after 4 h), with the majority clearing through the kidneys and intestines (48 % ID g−1 in the large intestine after 4 h).[164] Bioconjugates of [227Th][Th(Me-3,2-HOPO-OH)] have shown promise in the treatment of a variety of cancers in mouse models, including in the commencement of a Phase I clinical trial with PSMA (NCT03724747).[164,171–173]
The H8(3,4,3-LI(CAM)) chelator features four CAM functional groups.[166] The 232Th4+ complex has a formation constant of 1047.7. The kinetic inertness and radiolabelling with 227Th have not been reported.
The acyclic picolinate chelators H4octapa and H4py4pa have been investigated for use with Th4+.[160] The radiolabelling kinetics of H4octapa with 227Th were rapid, giving a radiolabelling efficiency of 65 % after 10 min at pH 5.5, which did not increase after 2.5 h. Radiolabelling of H4py4pa is slower under the same conditions, with 45 % radiolabelling efficiency after 10 min; however, this increased to 87 % after 2.5 h. Both complexes showed good inertness to PBS after 14 days at ambient temperature; however, stability under biologically relevant conditions has not yet been determined.
The high thermodynamic stability and kinetic inertness of thorium coordination complexes show significant promise for use in α-therapy. However, development of thorium-based radiopharmaceuticals requires a better understanding of the radiochemistry (e.g. optimised radiolabelling conditions) of 227Th as well as the in vivo stability of new complexes.
Radium
223Ra (Eα 5.7 MeV) and 224Ra (5.7 MeV) decay through the emission of four α-particles to the stable isotopes lead-207 (207Pb) and lead-208 (208Pb), respectively. Both isotopes have applications in α-therapy, and 223Ra also has a γ-ray emission that could be used in SPECT imaging, allowing the use of 223Ra as a theranostic.[153] Ra2+ acts similarly to Ca2+ in vivo, selectively absorbed by bone and areas of high metabolic activity such as cancerous tissue.[174] Ra2+ is cleared from the body through the intestinal tract, and the fast rate of clearance paired with the slow decay rate of the radionuclides reduces the non-specific dose to the patient.
Radium-223/224 Production
223Ra is readily available due to pre-existing infrastructure in place for the use of [223Ra][RaCl2] in clinical settings.[1] The parent isotope, 227Ac, is produced by neutron irradiation of naturally occurring radium-226 (226Ra). The availability of 224Ra is lower, as it is sourced from the 232Th decay chain found within stores of nuclear waste.
Radium Chemistry
Radium is always found in the +2 oxidation state and has an ionic radius of 1.48 Å (CN 8).[12] Ra2+ exhibits highly basic character and therefore, like other alkaline earth metal cations, only forms weak coordination complexes.[175] As a result, most radium compounds are simple ionic salts.[176] The radioactivity and toxicity of Ra2+ have limited studies of the chemical properties of the metal ion. To assist with these investigations, Ba2+ is often used as a non-radioactive congener. The aqueous chemistry of Ra2+ is defined by interactions with ions commonly found in higher concentrations, such as Ba2+, co-precipitating as Ba(Ra)SO4. However, Ra2+ forms the hydroxide Ra(OH)2 in aqueous solutions where co-precipitating ions are not present.[177]
Radium Chelators for Radiopharmaceuticals
The formation of weak coordination complexes with conventional chelators prompted an investigation of palladium-based polyoxometalates (POMs) for 223/224Ra, which showed simultaneous encapsulation and surface-bound incorporation of 224Ra.[181] The polyoxopalladates were able to be radiolabelled with 223Ra and the daughter isotopes in a one-pot preparation (80°C, 90 min) and remained 87.5 % intact after incubation in aqueous media for 96 h. The radiolabelled polyoxopalladates showed a high affinity to serum proteins, and in vivo stability experiments are still required to determine their viability for radiopharmaceutical applications.
Only a few select chelators have been investigated for use with Ra2+ radiopharmaceuticals (Table 6). Crown ethers are known to be useful candidates for chelating alkaline earth metal cations. Combination crown ethers and calixarenes are scaffolds that have been investigated for both 131Ba2+ and 223/224Ra2+.[182–184] The formation constant of the hydroxy derivative was determined to be 104.6 with Ba2+. Radiolabelling of the chelators with 223Ra has yet to be studied, and the solubility of these complexes in aqueous media remains a barrier for therapeutic applications.

|
H2macropa forms an 11-coordinate complex with Ba2+ through the N4O6 donor atoms of the ligand and oxygen from a solvent molecule (Fig. 21).[178] The solid-state structure is a racemic mixture of the Δ(δ,λ,δ)(δ,λ,δ) and Λ(λ,δ,λ)(λ,δ,λ) enantiomers.
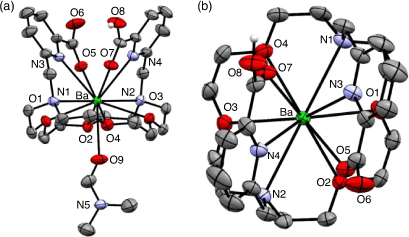
|
Radiolabelling with 131Ba (t1/2 11.5 days, γ 123.8 keV, 30 %) proceeded over 1 h at RT and pH 6 at concentrations above 10−4 M to achieve radiolabelling efficiency >95 %.[179] Radiolabelling of H2macropa with 223Ra2+ proceeded in 5 min at ambient temperature and physiological pH (7.4), giving >80 % radiolabelling efficiency at concentrations >10−5 M.[180,185] The high purity of [223Ra][Ra(Hmacropa)]+ eliminates the need for further purification before in vivo administration. The [223Ra][Ra(Hmacropa)]+ complex was stable in buffer and human serum at 37°C, with 90 % remaining intact over 12 days.[180] Biodistribution of [223Ra][Ra(Hmacropa)]+ indicated decreased bone uptake (1.6 % ID g−1 after 24 h) in vivo in a healthy rodent model when compared with [223Ra][RaCl2] (22 % ID g−1 after 24 h), demonstrating the absence of free 223Ra in circulation.[180] The similarity between the biodistributions of the H2macropa 223Ra and 131Ba complexes supports the use of 131Ba as a matched pair imaging agent for 223Ra α-therapy applications. H2macropa functionalised with β-alanine and the PSMA binding small molecule (((S)-5-tert-butoxy-4-(3-((S)-1,5-di-tert-butoxy-1,5-dioxopentan-2-yl)ureido)-5-oxopentanoic acid) (DUPA)) were radiolabelled with 223Ra under the same conditions as H2macropa with radiolabelling efficiency >90 %.[180] The radiolabelled conjugates [223Ra][Ra(macropa-β-alanine)] and [223Ra][Ra(macropa–DUPA)] remained >90 % and 75 % intact after 12 days in human serum at 37°C, respectively. [223Ra][Ra(macropa-β-alanine)] showed a similar biodistribution pattern in mice to [223Ra][Ra(Hmacropa)]+ with low bone uptake after 24 h (2.7 % ID g−1). Unexpectedly, [223Ra][Ra(macropa–DUPA)] exhibited no difference in biodistribution in mice compared with [223Ra][RaCl2], indicating that the construct was unstable in vivo and highlighting the significant impact that targeting vectors can have on the chemical properties and biodistribution.
The widespread availability of 223Ra and well-understood biodistribution in vivo makes it an appealing isotope for use in α-therapy. Indeed, it remains the only clinically approved α-emitting radiometal. New insights into the coordination chemistry of Ra2+ are removing the barriers in the development of 223Ra radiopharmaceuticals.
Recoil Nuclides and Nanoparticles
One of the problems that remains to be solved with chelators for use in α-therapy is the potential for uncontrolled redistribution of recoil daughter nuclides (and their potential α- or β-emitting progeny) leading to irradiation of healthy tissue.[2] The recoil energy released on α-decay (~100–200 keV) is at least 10 times higher than that required to break the coordination bonds (~10 eV) within the complex and can cause both loss of the radiometal and significant radiochemical degradation to the conjugated biomolecule due to the formation of radicals.[108,186,187] Once the daughter nuclides are released, re-complexation is unlikely because of competing factors within the biological milieu such as competing ligands and ions and the ultra-trace amounts of the chelator.[188] The recoil release is of greater importance for nuclides such as 225Ac, 223Ra, 224Ra, and 227Th that produce multiple α-emitting daughter radionuclides.[9] One possible concept for handling daughter isotopes is the use of encapsulating nanoparticles that allow for further surface chemistry for targeting (Table 7).[176]

|
Several studies have focussed on the immobilisation of 223/224Ra onto the surface of nanoparticles or incorporation into the crystalline structure as a way of delivering radiation to a target in vivo. The natural co-precipitation of 224Ra2+ with BaSO4 has been used as a template for incorporating 224Ra2+ into a crystalline BaSO4 nanoparticle structure.[191,192] The BaSO4 nanoparticles achieved 30–40 % radiolabelling efficiency with 224Ra.[191] The radiolabelled construct remained intact after treatment with ultrasound and incubation in deionised water for 7 days.
Hydroxyapatite (HAP) nanoparticles have been radiolabelled both through attachment of 223Ra to the surface of the nanoparticle and incorporation into the structure.[193] Radiolabelling yields >90 % were achieved at pH >7, which was attributed to the increased stability of surface interactions with 223Ra. Encouragingly, only a very small proportion of leaching of either 223Ra or the daughter radionuclide 221Bi and 211Pb was observed.[174] This was thought to be a result of the resorption of liberated radionuclides back into the nanoparticle structure, which is a potential advantage over more conventional chelator constructs.[194]
A method was recently developed using sodium nanozeolites whereby bioconjugation to a PSMA-targeting motif was demonstrated with a 223Ra labelled nanoparticle.[190] The sodium nanozeolites were radiolabelled with 223Ra by displacement of sodium, resulting in a 99.8 % radiolabelling efficiency. The surface was modified to add a silane-PEG modified linker and conjugated the anti-PSMA antibody D2B as a targeting moiety. The radiolabelled construct was stable, with more than 95 % remaining intact in human serum after 13 days.
Barium ferrite nanoparticles were radiolabelled with 223Ra using ionic displacement of Ba2+ cations and demonstrated a radiolabelling efficiency of 61 % ± 1.8 %.[175] The radiolabelled nanoparticles were conjugated to trastuzumab and demonstrated affinity, internalisation, and high cytotoxicity towards human ovarian adenocarcinoma SKOV-3 cells overexpressing HER2 receptor. The construct almost quantitatively retained 223Ra, 211Bi, and 211Pb, indicating potential for this method as means to control the biodistribution of 223Ra and the daughter nuclides in vivo.
Although the use of nanoparticles with 225Ac is fairly new, gold, silica, and iron oxide particles have all been recently radiolabelled with 225Ac and demonstrated a cytotoxic response in vivo.[195–197] The iron oxide particles demonstrated the capacity to retain 90 % of the daughter nuclides 221Fr and 213Bi in saline solution but this decreased to 70 % in human serum after 10 days. Intravenous administration of the radiobioconjugate in tumour-bearing mice demonstrated one of the shortcomings of nanoparticles, namely the large size causing rapid blood clearance, high accumulation of radiation in the liver, lungs, and spleen and very low tumour accumulation.[197]
Conclusions
Synthetic coordination chemistry is playing a prominent role in the development of new radiopharmaceuticals that aim to realise the potential of the medically useful α-emitting radiometals. For most of these radiometals, the most frequently used chelator has been H4DOTA, which has resulted in these complexes having fairly well understood coordination chemistry and radiochemistry. Such indiscriminate use of H4DOTA, however, ignores the varied fundamental characteristics of metal ions such as charge and ionic radius and the distinct preferences each has for geometry, CN, and donor atoms.
The development of new chelators using macrocycles based on cyclen, crown ethers, bispidine, and [2.2.2]-cryptand is addressing many of the issues associated with H4DOTA such as slow radiolabelling kinetics. In addition, new multidentate acyclic chelators are proving as kinetically inert as the macrocyclic counterparts. Many of these chelators have progressed to being bifunctionalised to generate radiopharmaceuticals with tumour-targeting functionality and are being evaluated in vivo.
The in vivo toxicity related to the α-emitting radionuclides as well as released recoil progeny are a difficult challenge, with kidneys often the dose-limiting organs. In addition to the use of nanoparticles, rapid internalisation and retention in tumours could be a strategic method to prevent recoil products causing considerable toxicity to healthy tissues. Possessing chelators with a range of different properties is beneficial to be able to manipulate the biodistribution, tumour uptake, and retention of radiopharmaceuticals.
The progression of radiopharmaceuticals for TAT from research to the clinic is a challenging proposition. Important issues include the availability of radionuclides as well as the infrastructure to produce and handle radionuclides in a safe and economically viable manner. Success will depend on providing solutions to the availability and handling issues problems as much as practical and elegant coordination chemistry is providing solutions to the problems of radiolabelling efficiency, stability, selectivity, and toxicity.
Data Availability Statement
Data sharing is not applicable as no new data were generated or analysed during this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
We acknowledge the Australian Government for support with a National Imaging Facility Fellowship (NCRIS) and Research Training Program (RTP) Scholarship.
References
[1] E. Deshayes, M. Roumiguie, C. Thibault, P. Beuzeboc, F. Cachin, C. Hennequin, D. Huglo, F. Rozet, D. Kassab-Chahmi, X. Rebillard, N. Houede, Drug Des. Devel. Ther. 2017, 11, 2643.| Crossref | GoogleScholarGoogle Scholar | 28919714PubMed |
[2] R. M. de Kruijff, H. T. Wolterbeek, A. G. Denkova, Pharmaceuticals 2015, 8, 321.
| Crossref | GoogleScholarGoogle Scholar | 26066613PubMed |
[3] S. Poty, L. C. Francesconi, M. R. McDevitt, M. J. Morris, J. S. Lewis, J. Nucl. Med. 2018, 59, 878.
| Crossref | GoogleScholarGoogle Scholar | 29545378PubMed |
[4] M. G. Ferrier, V. Radchenko, D. S. Wilbur, Radiochim. Acta 2019, 107, 1065.
| Crossref | GoogleScholarGoogle Scholar |
[5] C. Keller, W. Wolf, J. Shani, in Ullmann’s Encyclopedia of Industrial Chemistry (Ed. C. Ley) 2012, pp. 89–117 (Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim).
[6] K. Fujiki, Y. Kanayama, S. Yano, N. Sato, T. Yokokita, P. Ahmadi, Y. Watanabe, H. Haba, K. Tanaka, Chem. Sci. 2019, 10, 1936.
| Crossref | GoogleScholarGoogle Scholar | 30881623PubMed |
[7] A. Morgenstern, C. Apostolidis, F. Bruchertseifer, Semin. Nucl. Med. 2020, 50, 119.
| Crossref | GoogleScholarGoogle Scholar | 32172796PubMed |
[8] R. A. Boll, D. Malkemus, S. Mirzadeh, Appl. Radiat. Isot. 2005, 62, 667.
| Crossref | GoogleScholarGoogle Scholar | 15763472PubMed |
[9] B. J. B. Nelson, J. D. Andersson, F. Wuest, Pharmaceutics 2021, 13, 49.
| Crossref | GoogleScholarGoogle Scholar |
[10] J. R. Griswold, D. G. Medvedev, J. W. Engle, R. Copping, J. M. Fitzsimmons, V. Radchenko, J. C. Cooley, M. E. Fassbender, D. L. Denton, K. E. Murphy, A. C. Owens, E. R. Birnbaum, K. D. John, F. M. Nortier, D. W. Stracener, L. H. Heilbronn, L. F. Mausner, S. Mirzadeh, Appl. Radiat. Isot. 2016, 118, 366.
| Crossref | GoogleScholarGoogle Scholar | 27776333PubMed |
[11] N. A. Thiele, J. J. Wilson, Cancer Biother. Radiopharm. 2018, 33, 336.
| Crossref | GoogleScholarGoogle Scholar | 29889562PubMed |
[12] R. Shannon, Acta Crystallogr. A 1976, 32, 751.
| Crossref | GoogleScholarGoogle Scholar |
[13] G. J. P. Deblonde, M. Zavarin, A. B. Kersting, Coord. Chem. Rev. 2021, 446, 214130.
| Crossref | GoogleScholarGoogle Scholar |
[14] N. A. Thiele, V. Brown, J. M. Kelly, A. Amor-Coarasa, U. Jermilova, S. N. MacMillan, A. Nikolopoulou, S. Ponnala, C. F. Ramogida, A. K. H. Robertson, C. Rodriguez-Rodriguez, P. Schaffer, C. Williams, J. W. Babich, V. Radchenko, J. J. Wilson, Angew. Chem. Int. Ed. 2017, 56, 14712.
| Crossref | GoogleScholarGoogle Scholar |
[15] R. G. Pearson, J. Chem. Educ. 1968, 45, 581.
| Crossref | GoogleScholarGoogle Scholar |
[16] R. G. Pearson, J. Am. Chem. Soc. 1963, 85, 3533.
| Crossref | GoogleScholarGoogle Scholar |
[17] S. A. Cotton, C. R. Chim. 2005, 8, 129.
| Crossref | GoogleScholarGoogle Scholar |
[18] M. G. Ferrier, B. W. Stein, E. R. Batista, J. M. Berg, E. R. Birnbaum, J. W. Engle, K. D. John, S. A. Kozimor, J. S. Lezama Pacheco, L. N. Redman, ACS Cent. Sci. 2017, 3, 176.
| Crossref | GoogleScholarGoogle Scholar | 28386595PubMed |
[19] I. A. Davis, K. A. Glowienka, R. A. Boll, K. A. Deal, M. W. Brechbiel, M. Stabin, P. N. Bochsler, S. Mirzadeh, S. J. Kennel, Nucl. Med. Biol. 1999, 26, 581.
| Crossref | GoogleScholarGoogle Scholar | 10473198PubMed |
[20] L. Wharton, E. Kurakina, V. Radchenko, P. Schaffer, C. Orvig, Inorg. Chem. 2021, 60, 4076.
| Crossref | GoogleScholarGoogle Scholar | 33635057PubMed |
[21] C. F. Ramogida, A. K. H. Robertson, U. Jermilova, C. Zhang, H. Yang, P. Kunz, J. Lassen, I. Bratanovic, V. Brown, L. Southcott, C. Rodriguez-Rodriguez, V. Radchenko, F. Benard, C. Orvig, P. Schaffer, EJNMMI Radiopharm. Chem. 2019, 4, 21.
| Crossref | GoogleScholarGoogle Scholar | 31659557PubMed |
[22] L. Li, J. Rousseau, M. G. Jaraquemada-Pelaez, X. Wang, A. Robertson, V. Radchenko, P. Schaffer, K. S. Lin, F. Benard, C. Orvig, Bioconjug. Chem. 2021, 32, 1348.
| Crossref | GoogleScholarGoogle Scholar | 32216377PubMed |
[23] P. Comba, U. Jermilova, C. Orvig, B. O. Patrick, C. F. Ramogida, K. Ruck, C. Schneider, M. Starke, Chem. – Eur. J. 2017, 23, 15945.
| Crossref | GoogleScholarGoogle Scholar | 28815804PubMed |
[24] B. W. Stein, A. Morgenstern, E. R. Batista, E. R. Birnbaum, S. E. Bone, S. K. Cary, M. G. Ferrier, K. D. John, J. L. Pacheco, S. A. Kozimor, V. Mocko, B. L. Scott, P. Yang, J. Am. Chem. Soc. 2019, 141, 19404.
| Crossref | GoogleScholarGoogle Scholar | 31794205PubMed |
[25] F. Reissig, D. Bauer, K. Zarschler, Z. Novy, K. Bendova, M.-C. Ludik, K. Kopka, H.-J. Pietzsch, M. Petrik, C. Mamat, Cancers 2021, 13, 1974.
| Crossref | GoogleScholarGoogle Scholar | 33923965PubMed |
[26] P. Garnuszek, U. Karczmarczyk, M. Maurin, A. Sikora, J. Zaborniak, J. Pijarowska-Kruszyna, A. Jaroń, M. Wyczółkowska, W. Wojdowska, D. Pawlak, P. F. J. Lipiński, R. Mikołajczak, Int. J. Mol. Sci. 2021, 22, 2731.
| Crossref | GoogleScholarGoogle Scholar | 33800517PubMed |
[27] B. Feuerecker, R. Tauber, K. Knorr, M. Heck, A. Beheshti, C. Seidl, F. Bruchertseifer, A. Pickhard, A. Gafita, C. Kratochwil, M. Retz, J. E. Gschwend, W. A. Weber, C. D’Alessandria, A. Morgenstern, M. Eiber, Eur. Urol. 2021, 79, 343.
| Crossref | GoogleScholarGoogle Scholar | 33293081PubMed |
[28] A. D. Stuparu, C. A. L. Meyer, S. L. Evans-Axelsson, K. Lückerath, L. H. Wei, W. Kim, S. Poddar, C. E. Mona, M. Dahlbom, M. D. Girgis, C. G. Radu, J. Czernin, R. Slavik, Theranostics 2020, 10, 2612.
| Crossref | GoogleScholarGoogle Scholar | 32194823PubMed |
[29] M. Sathekge, F. Bruchertseifer, M. Vorster, I. O. Lawal, O. Knoesen, J. Mahapane, C. Davis, F. Reyneke, A. Maes, C. Kratochwil, T. Lengana, F. L. Giesel, C. Van de Wiele, A. Morgenstern, J. Nucl. Med. 2020, 61, 62.
| Crossref | GoogleScholarGoogle Scholar | 31101746PubMed |
[30] C. Kratochwil, F. Bruchertseifer, F. L. Giesel, M. Weis, F. A. Verburg, F. Mottaghy, K. Kopka, C. Apostolidis, U. Haberkorn, A. Morgenstern, J. Nucl. Med. 2016, 57, 1941.
| Crossref | GoogleScholarGoogle Scholar | 27390158PubMed |
[31] S. Poty, R. Membreno, J. M. Glaser, A. Ragupathi, W. W. Scholz, B. M. Zeglis, J. S. Lewis, Chem. Commun. 2018, 2599.
| Crossref | GoogleScholarGoogle Scholar |
[32] A. Kovacs, ACS Omega 2020, 5, 26431.
| Crossref | GoogleScholarGoogle Scholar | 33110971PubMed |
[33] A. Kovács, Z. Varga, Struct. Chem. 2021, 32, 643.
| Crossref | GoogleScholarGoogle Scholar |
[34] J. M. Kelly, A. Amor-Coarasa, S. Ponnala, A. Nikolopoulou, C. Williams, N. A. Thiele, D. Schlyer, J. J. Wilson, S. G. DiMagno, J. W. Babich, J. Nucl. Med. 2019, 60, 649.
| Crossref | GoogleScholarGoogle Scholar | 30413660PubMed |
[35] H. Yang, C. Zhang, Z. Yuan, C. Rodriguez-Rodriguez, A. Robertson, V. Radchenko, R. Perron, D. Gendron, P. Causey, F. Gao, F. Benard, P. Schaffer, Chem. – Eur. J. 2020, 26, 11435.
| Crossref | GoogleScholarGoogle Scholar | 32588455PubMed |
[36] S. Aime, A. Barge, F. Benetollo, G. Bombieri, M. Botta, F. Uggeri, Inorg. Chem. 1997, 36, 4287.
| Crossref | GoogleScholarGoogle Scholar |
[37] J. Rohovec, P. Vojtíšek, P. Hermann, J. Mosinger, Z. Žák, I. Lukeš, Dalton Trans. 1999, 3585.
| Crossref | GoogleScholarGoogle Scholar |
[38] S. Aime, M. Botta, M. Fasano, M. P. M. Marques, C. F. G. C. Geraldes, D. Pubanz, A. E. Merbach, Inorg. Chem. 1997, 36, 2059.
| Crossref | GoogleScholarGoogle Scholar | 11669824PubMed |
[39] V. R. Solomon, E. Alizadeh, W. Bernhard, S. V. Hartimath, W. Hill, R. Chekol, K. M. Barreto, C. R. Geyer, H. Fonge, Mol. Pharm. 2019, 16, 4807.
| Crossref | GoogleScholarGoogle Scholar | 31518138PubMed |
[40] K. A. Deal, I. A. Davis, S. Mirzadeh, S. J. Kennel, M. W. Brechbiel, J. Med. Chem. 1999, 42, 2988.
| Crossref | GoogleScholarGoogle Scholar | 10425108PubMed |
[41] L. Królicki, F. Bruchertseifer, J. Kunikowska, H. Koziara, D. Pawlak, R. Kuliński, R. Rola, A. Merlo, A. Morgenstern, Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3595.
| Crossref | GoogleScholarGoogle Scholar | 33860346PubMed |
[42] Y. Qin, S. Imobersteg, A. Blanc, S. Frank, R. Schibli, M. P. Béhé, M. Grzmil, Pharmaceutics 2020, 12, 1088.
| Crossref | GoogleScholarGoogle Scholar |
[43] V. J. Kelly, S.-T. Wu, V. Gottumukkala, R. Coelho, K. Palmer, S. Nair, T. Erick, R. Puri, O. Ilovich, P. Mukherjee, Theranostics 2020, 10, 6946.
| Crossref | GoogleScholarGoogle Scholar | 32550914PubMed |
[44] K. Kamaleshwaran, M. Suneelkumar, R. Madhusairam, E. Radhakrishnan, S. Arunpandiyan, V. Arnold, Indian J. Nucl. Med. 2020, 35, 226.
| Crossref | GoogleScholarGoogle Scholar | 33082679PubMed |
[45] A. Cortez, A. Josefsson, G. McCarty, A. E. Shtekler, A. Rao, Z. Austin, J. R. Nedrow, Nucl. Med. Biol. 2020, 88–89, 62.
| Crossref | GoogleScholarGoogle Scholar | 32799049PubMed |
[46] N. Pfannkuchen, N. Bausbacher, S. Pektor, M. Miederer, F. Rosch, Curr. Radiopharm. 2018, 11, 223.
| Crossref | GoogleScholarGoogle Scholar | 29866026PubMed |
[47] D. N. Pandya, R. Hantgan, M. M. Budzevich, N. D. Kock, D. L. Morse, I. Batista, A. Mintz, K. C. Li, T. J. Wadas, Theranostics 2016, 6, 698.
| Crossref | GoogleScholarGoogle Scholar | 27022417PubMed |
[48] M. R. McDevitt, D. Ma, J. Simon, R. K. Frank, D. A. Scheinberg, Appl. Radiat. Isot. 2002, 57, 841.
| Crossref | GoogleScholarGoogle Scholar | 12406626PubMed |
[49] M. R. McDevitt, D. Ma, L. T. Lai, J. Simon, P. Borchardt, R. K. Frank, K. Wu, V. Pellegrini, M. J. Curcio, M. Miederer, N. H. Bander, D. A. Scheinberg, Science 2001, 294, 1537.
| Crossref | GoogleScholarGoogle Scholar | 11711678PubMed |
[50] M. Altai, R. Membreno, B. Cook, V. Tolmachev, B. M. Zeglis, J. Nucl. Med. 2017, 58, 1553.
| Crossref | GoogleScholarGoogle Scholar | 28687600PubMed |
[51] H. Rathke, F. Bruchertseifer, C. Kratochwil, H. Keller, F. L. Giesel, C. Apostolidis, U. Haberkorn, A. Morgenstern, Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 311.
| Crossref | GoogleScholarGoogle Scholar | 32468252PubMed |
[52] A. Hu, I. Keresztes, S. N. MacMillan, Y. Yang, E. Ding, W. R. Zipfel, R. A. DiStasio, J. W. Babich, J. J. Wilson, Inorg. Chem. 2020, 59, 5116.
| Crossref | GoogleScholarGoogle Scholar | 32216281PubMed |
[53] A. Roca-Sabio, M. Mato-Iglesias, D. Esteban-Gómez, É. Tóth, A. d. Blas, C. Platas-Iglesias, T. Rodríguez-Blas, J. Am. Chem. Soc. 2009, 131, 3331.
| Crossref | GoogleScholarGoogle Scholar | 19256570PubMed |
[54] E. J. Corey, J. C. Bailar, J. Am. Chem. Soc. 1959, 81, 2620.
| Crossref | GoogleScholarGoogle Scholar |
[55] A. Hu, S. N. MacMillan, J. J. Wilson, J. Am. Chem. Soc. 2020, 142, 13500.
| Crossref | GoogleScholarGoogle Scholar | 32697907PubMed |
[56] A. Morgenstern, L. M. Lilley, B. W. Stein, S. A. Kozimor, E. R. Batista, P. Yang, Inorg. Chem. 2021, 60, 623.
| Crossref | GoogleScholarGoogle Scholar | 33213142PubMed |
[57] H. Yang, C. Zhang, Z. Yuan, C. Rodriguez-Rodriguez, A. Robertson, V. Radchenko, R. Perron, D. Gendron, P. Causey, F. Gao, F. Bénard, P. Schaffer, Chem. – Eur. J. 2020, 26, 11435.
| Crossref | GoogleScholarGoogle Scholar | 32588455PubMed |
[58] J. Yang, J. Xu, L. Cheuy, R. Gonzalez, D. R. Fisher, Y. Miao, Mol. Pharm. 2019, 16, 1694.
| Crossref | GoogleScholarGoogle Scholar | 30763112PubMed |
[59] A.-M. Frelin-Labalme, T. Roger, N. Falzone, B. Quan Lee, N. R. Sibson, K. A. Vallis, M. Bernaudin, S. Valable, A. Corroyer-Dulmont, Med. Phys. 2020, 47, 1317.
| Crossref | GoogleScholarGoogle Scholar | 31838744PubMed |
[60] M. Sathekge, O. Knoesen, M. Meckel, M. Modiselle, M. Vorster, S. Marx, Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1099.
| Crossref | GoogleScholarGoogle Scholar | 28255795PubMed |
[61] H. A. Song, C. S. Kang, K. E. Baidoo, D. E. Milenic, Y. Chen, A. Dai, M. W. Brechbiel, H. S. Chong, Bioconjug. Chem. 2011, 22, 1128.
| Crossref | GoogleScholarGoogle Scholar | 21604692PubMed |
[62] D. Wild, M. Frischknecht, H. Zhang, A. Morgenstern, F. Bruchertseifer, J. Boisclair, A. Provencher-Bolliger, J.-C. Reubi, H. R. Maecke, Cancer Res. 2011, 71, 1009.
| Crossref | GoogleScholarGoogle Scholar | 21245097PubMed |
[63] L. I. Guseva, Radiochemistry 2014, 56, 451.
| Crossref | GoogleScholarGoogle Scholar |
[64] S. Ahenkorah, I. Cassells, C. M. Deroose, T. Cardinaels, A. R. Burgoyne, G. Bormans, M. Ooms, F. Cleeren, Pharmaceutics 2021, 13, 599.
| Crossref | GoogleScholarGoogle Scholar | 33919391PubMed |
[65] G. Sgouros, A. M. Ballangrud, J. G. Jurcic, M. R. McDevitt, J. L. Humm, Y. E. Erdi, B. M. Mehta, R. D. Finn, S. M. Larson, D. A. Scheinberg, J. Nucl. Med. 1999, 40, 1935.
| Crossref | GoogleScholarGoogle Scholar | 10565792PubMed |
[66] J. de Swart, H. S. Chan, M. C. Goorden, A. Morgenstern, F. Bruchertseifer, F. J. Beekman, M. de Jong, M. W. Konijnenberg, J. Nucl. Med. 2016, 57, 486.
| Crossref | GoogleScholarGoogle Scholar | 26635343PubMed |
[67] P. Manna, D. Szücs, T. Csupász, A. Fekete, D. Szikra, Z. Lin, A. Gáspár, S. Bhattacharya, A. Zulaica, I. Tóth, U. Kortz, Inorg. Chem. 2020, 59, 16769.
| Crossref | GoogleScholarGoogle Scholar | 33174740PubMed |
[68] F. Bruchertseifer, A. Kellerbauer, R. Malmbeck, A. Morgenstern, J. Labelled. Compd. Radiopharm. 2019, 62, 794.
| Crossref | GoogleScholarGoogle Scholar |
[69] A. Morgenstern, C. Apostolidis, C. Kratochwil, M. Sathekge, L. Krolicki, F. Bruchertseifer, Curr. Radiopharm. 2018, 11, 200.
| Crossref | GoogleScholarGoogle Scholar | 29732998PubMed |
[70] M. R. McDevitt, R. D. Finn, G. Sgouros, D. Ma, D. A. Scheinberg, Appl. Radiat. Isot. 1999, 50, 895.
| Crossref | GoogleScholarGoogle Scholar | 10214708PubMed |
[71] R. Pujales-Paradela, A. Rodríguez-Rodríguez, A. Gayoso-Padula, I. Brandariz, L. Valencia, D. Esteban-Gómez, C. Platas-Iglesias, Dalton Trans. 2018, 47, 13830.
| Crossref | GoogleScholarGoogle Scholar | 30230496PubMed |
[72] H. Sun, H. Li, P. J. Sadler, Chem. Ber. 1997, 130, 669.
| Crossref | GoogleScholarGoogle Scholar |
[73] H. S. Chan, E. de Blois, M. W. Konijnenberg, A. Morgenstern, F. Bruchertseifer, J. P. Norenberg, F. J. Verzijlbergen, M. de Jong, W. A. P. Breeman, EJNMMI Radiopharm. Chem. 2017, 1, 9.
| Crossref | GoogleScholarGoogle Scholar | 29564386PubMed |
[74] G. G. Briand, N. Burford, Chem. Rev. 1999, 99, 2601.
| Crossref | GoogleScholarGoogle Scholar | 11749495PubMed |
[75] P. J. Sadler, H. Sun, H. Li, Chemistry 1996, 2, 701.
| Crossref | GoogleScholarGoogle Scholar |
[76] M.-X. Li, M. Yang, J.-Y. Niu, L.-Z. Zhang, S.-Q. Xie, Inorg. Chem. 2012, 51, 12521.
| Crossref | GoogleScholarGoogle Scholar | 23136979PubMed |
[77] L. Dorso, E. Bigot-Corbel, J. Abadie, M. Diab, S. Gouard, F. Bruchertseifer, A. Morgenstern, C. Maurel, M. Chérel, F. Davodeau, PLoS One 2016, 11, e0151330.
| Crossref | GoogleScholarGoogle Scholar | 26982495PubMed |
[78] K. Garmestani, Z. Yao, M. Zhang, K. Wong, C. W. Park, I. Pastan, J. A. Carrasquillo, M. W. Brechbiel, Nucl. Med. Biol. 2001, 28, 409.
| Crossref | GoogleScholarGoogle Scholar | 11395314PubMed |
[79] D. E. Milenic, M. Roselli, S. Mirzadeh, C. G. Pippin, O. A. Gansow, D. Colcher, M. W. Brechbiel, J. Schlom, Cancer Biother. Radiopharm. 2001, 16, 133.
| Crossref | GoogleScholarGoogle Scholar | 11385960PubMed |
[80] J. Schwartz, J. S. Jaggi, J. A. O’Donoghue, S. Ruan, M. McDevitt, S. M. Larson, D. A. Scheinberg, J. L. Humm, Phys. Med. Biol. 2011, 56, 721.
| Crossref | GoogleScholarGoogle Scholar | 21220845PubMed |
[81] S. P. Summers, K. A. Abboud, S. R. Farrah, G. J. Palenik, Inorg. Chem. 1994, 33, 88.
| Crossref | GoogleScholarGoogle Scholar |
[82] M. W. Brechbiel, O. A. Gansow, C. G. Pippin, R. D. Rogers, R. P. Planalp, Inorg. Chem. 1996, 35, 6343.
| Crossref | GoogleScholarGoogle Scholar |
[83] T. K. Nikula, M. R. McDevitt, R. D. Finn, C. Wu, R. W. Kozak, K. Garmestani, M. W. Brechbiel, M. J. Curcio, C. G. Pippin, L. Tiffany-Jones, M. W. Geerlings, C. Apostolidis, R. Molinet, M. W. Geerlings, O. A. Gansow, D. A. Scheinberg, J. Nucl. Med. 1999, 40, 166.
| 9935073PubMed |
[84] F. Bruchertseifer, P. Comba, B. Martin, A. Morgenstern, J. Notni, M. Starke, H. Wadepohl, ChemMedChem 2020, 15, 1591.
| Crossref | GoogleScholarGoogle Scholar | 32613737PubMed |
[85] D. J. Fiszbein, V. Brown, N. A. Thiele, J. J. Woods, L. Wharton, S. N. MacMillan, V. Radchenko, C. F. Ramogida, J. J. Wilson, Inorg. Chem. 2021, 60, 9199.
| Crossref | GoogleScholarGoogle Scholar | 34102841PubMed |
[86] E. Csajbok, Z. Baranyai, I. Banyai, E. Brucher, R. Kiraly, A. Muller-Fahrnow, J. Platzek, B. Raduchel, M. Schafer, Inorg. Chem. 2003, 42, 2342.
| Crossref | GoogleScholarGoogle Scholar | 12665368PubMed |
[87] J. Simecek, P. Hermann, C. Seidl, F. Bruchertseifer, A. Morgenstern, H. J. Wester, J. Notni, EJNMMI Res. 2018, 8, 78.
| Crossref | GoogleScholarGoogle Scholar | 30091088PubMed |
[88] B. Bartos, K. Lyczko, A. Kasperek, S. Krajewski, A. Bilewicz, J. Radioanal. Nucl. Chem. 2013, 295, 205.
| Crossref | GoogleScholarGoogle Scholar | 26224928PubMed |
[89] D. Horváth, F. Travagin, N. Guidolin, F. Buonsanti, G. Tircsó, I. Tóth, F. Bruchertseifer, A. Morgenstern, J. Notni, G. B. Giovenzana, Z. Baranyai, Inorg. Chem. Front. 2021, 8, 3893.
| Crossref | GoogleScholarGoogle Scholar |
[90] J. J. Wilson, M. Ferrier, V. Radchenko, J. R. Maassen, J. W. Engle, E. R. Batista, R. L. Martin, F. M. Nortier, M. E. Fassbender, K. D. John, E. R. Birnbaum, Nucl. Med. Biol. 2015, 42, 428.
| Crossref | GoogleScholarGoogle Scholar | 25684650PubMed |
[91] L. M. Lima, M. Beyler, F. Oukhatar, P. Le Saec, A. Faivre-Chauvet, C. Platas-Iglesias, R. Delgado, R. Tripier, Chem. Commun. 2014, 12371.
| Crossref | GoogleScholarGoogle Scholar |
[92] J. L. Lange, P. R. W. J. Davey, M. T. Ma, J. M. White, A. Morgenstern, F. Bruchertseifer, P. J. Blower, B. M. Paterson, Dalton Trans. 2020, 49, 14962.
| Crossref | GoogleScholarGoogle Scholar | 33079111PubMed |
[93] A. E. Martell, R. M. Smith, Critical Stability Constants 1974 (Plenum Press: New York, NY).
[94] M. W. Brechbiel, O. A. Gansow, J. Chem. Soc., Perkin Trans. 1 1992, 1173.
| Crossref | GoogleScholarGoogle Scholar |
[95] S. Hassfjell, K. O. Kongshaug, C. Rømming, Dalton Trans. 2003, 1433.
| Crossref | GoogleScholarGoogle Scholar |
[96] K. Kumar, M. Magerstädt, O. A. Gansow, J. Chem. Soc. 1989, 145.
| Crossref | GoogleScholarGoogle Scholar |
[97] C. L. Ruegg, W. T. Anderson-Berg, M. W. Brechbiel, S. Mirzadeh, O. A. Gansow, M. Strand, Cancer Res. 1990, 50, 4221.
| 2364380PubMed |
[98] R. P. Junghans, D. Dobbs, M. W. Brechbiel, S. Mirzadeh, A. A. Raubitschek, O. A. Gansow, T. A. Waldmann, Cancer Res. 1993, 53, 5683.
| 8242624PubMed |
[99] C. Kratochwil, K. Schmidt, A. Afshar-Oromieh, F. Bruchertseifer, H. Rathke, A. Morgenstern, U. Haberkorn, F. L. Giesel, Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 31.
| Crossref | GoogleScholarGoogle Scholar | 28891033PubMed |
[100] C. Kratochwil, F. L. Giesel, F. Bruchertseifer, W. Mier, C. Apostolidis, R. Boll, K. Murphy, U. Haberkorn, A. Morgenstern, Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2106.
| Crossref | GoogleScholarGoogle Scholar | 25070685PubMed |
[101] M. Sathekge, O. Knoesen, M. Meckel, M. Modiselle, M. Vorster, S. Marx, Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1099.
| Crossref | GoogleScholarGoogle Scholar | 28255795PubMed |
[102] L. Marcu, E. Bezak, B. J. Allen, Crit. Rev. Oncol. Hematol. 2018, 123, 7.
| Crossref | GoogleScholarGoogle Scholar | 29482781PubMed |
[103] I. Lazar, D. C. Hrncir, W. D. Kim, G. E. Kiefer, A. D. Sherry, Inorg. Chem. 1992, 31, 4422.
| Crossref | GoogleScholarGoogle Scholar |
[104] C. S. Kang, H. A. Song, D. E. Milenic, K. E. Baidoo, M. W. Brechbiel, H. S. Chong, Nucl. Med. Biol. 2013, 40, 600.
| Crossref | GoogleScholarGoogle Scholar | 23541026PubMed |
[105] L. M. Lima, M. Beyler, R. Delgado, C. Platas-Iglesias, R. Tripier, Inorg. Chem. 2015, 54, 7045.
| Crossref | GoogleScholarGoogle Scholar | 26146022PubMed |
[106] K. Yong, M. W. Brechbiel, Dalton Trans. 2011, 6068.
| Crossref | GoogleScholarGoogle Scholar | 21380408PubMed |
[107] S. Mirzadeh, K. Kumar, O. A. Gansow, Radiochim. Acta 1993, 60, 1.
| Crossref | GoogleScholarGoogle Scholar |
[108] V. Y. Stenberg, A Juzeniene, Ø. S. Bruland, R. H. Larsen, Curr. Radiopharm. 2020, 13, 130.
| Crossref | GoogleScholarGoogle Scholar | 32389119PubMed |
[109] M. Pruszyński, R. Walczak, M. Rodak, F. Bruchertseifer, A. Morgenstern, A. Bilewicz, Appl. Radiat. Isot. 2021, 172, 109655.
| Crossref | GoogleScholarGoogle Scholar | 33657491PubMed |
[110] B. L. McNeil, A. K. H. Robertson, W. Fu, H. Yang, C. Hoehr, C. F. Ramogida, P. Schaffer, EJNMMI Radiopharm. Chem. 2021, 6, 6.
| Crossref | GoogleScholarGoogle Scholar | 33527221PubMed |
[111] D. Mathe, K. Szigeti, N. Hegedus, I. Horvath, D. S. Veres, B. Kovacs, Z. Szucs, Appl. Radiat. Isot. 2016, 114, 1.
| Crossref | GoogleScholarGoogle Scholar | 27156049PubMed |
[112] C. F. Baes, R. E. Mesmer, The Hydrolysis of Cations 1976 (Wiley: New York, NY).
[113] J. Parr, Polyhedron 1997, 16, 551.
| Crossref | GoogleScholarGoogle Scholar |
[114] A. Ingham, T. I. Kostelnik, B. L. McNeil, B. O. Patrick, N. Choudhary, M. G. Jaraquemada-Peláez, C. Orvig, Dalton Trans. 2021, 11579.
| Crossref | GoogleScholarGoogle Scholar | 34352061PubMed |
[115] J. W. Nugent, H.-S. Lee, J. H. Reibenspies, R. D. Hancock, Polyhedron 2015, 91, 120.
| Crossref | GoogleScholarGoogle Scholar |
[116] J. C. dos Santos, M. Schafer, U. Bauder-Wust, W. Lehnert, K. Leotta, A. Morgenstern, K. Kopka, U. Haberkorn, W. Mier, C. Kratochwil, Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1081.
| Crossref | GoogleScholarGoogle Scholar | 30603987PubMed |
[117] L. L. Chappell, E. Dadachova, D. E. Milenic, K. Garmestani, C. Wu, M. W. Brechbiel, Nucl. Med. Biol. 2000, 27, 93.
| Crossref | GoogleScholarGoogle Scholar | 10755652PubMed |
[118] F. Cuenot, M. Meyer, E. Espinosa, A. Bucaille, R. Burgat, R. Guilard, C. Marichal-Westrich, Eur. J. Inorg. Chem. 2008, 267.
| Crossref | GoogleScholarGoogle Scholar |
[119] S. R. Banerjee, I. Minn, V. Kumar, A. Josefsson, A. Lisok, M. Brummet, J. Chen, A. P. Kiess, K. Baidoo, C. Brayton, R. C. Mease, M. Brechbiel, G. Sgouros, R. F. Hobbs, M. G. Pomper, J. Nucl. Med. 2020, 61, 80.
| Crossref | GoogleScholarGoogle Scholar | 31253744PubMed |
[120] V. Y. Stenberg, A. Juzeniene, Q. Chen, X. Yang, O. S. Bruland, R. H. Larsen, J. Labelled Comp. Radiopharm. 2020, 63, 129.
| Crossref | GoogleScholarGoogle Scholar | 31919866PubMed |
[121] R. F. Meredith, J. J. Torgue, T. A. Rozgaja, E. P. Banaga, P. W. Bunch, R. D. Alvarez, J. M. Straughn, M. C. Dobelbower, A. M. Lowy, Am. J. Clin. Oncol. 2018, 41, 716.
| Crossref | GoogleScholarGoogle Scholar | 27906723PubMed |
[122] S. Westrom, R. Generalov, T. B. Bonsdorff, R. H. Larsen, Nucl. Med. Biol. 2017, 51, 1.
| Crossref | GoogleScholarGoogle Scholar | 28486098PubMed |
[123] T. A. R. Stallons, A. Saidi, I. Tworowska, E. S. Delpassand, J. J. Torgue, Mol. Cancer Ther. 2019, 18, 1012.
| Crossref | GoogleScholarGoogle Scholar |
[124] A. W. McDonagh, B. L. McNeil, B. O. Patrick, C. F. Ramogida, Inorg. Chem. 2021, 60, 10030.
| Crossref | GoogleScholarGoogle Scholar | 34159785PubMed |
[125] C. G. Pippin, T. J. McMurry, M. W. Brechbiel, M. McDonald, R. Lambrecht, D. Milenic, M. Roselli, D. Colcher, O. A. Gansow, Inorg. Chim. Acta 1995, 239, 43.
| Crossref | GoogleScholarGoogle Scholar |
[126] F. Cuenot, M. Meyer, E. Espinosa, A. Bucaille, R. Burgat, R. Guilard, C. Marichal-Westrich, Eur. J. Inorg. Chem. 2008, 267.
| Crossref | GoogleScholarGoogle Scholar |
[127] Y. Miao, T. P. Quinn, Crit. Rev. Oncol. Hematol. 2008, 67, 213.
| Crossref | GoogleScholarGoogle Scholar | 18387816PubMed |
[128] Y. Miao, M. Hylarides, D. R. Fisher, T. Shelton, H. Moore, D. W. Wester, A. R. Fritzberg, C. T. Winkelmann, T. Hoffman, T. P. Quinn, Clin. Cancer Res. 2005, 11, 5616.
| Crossref | GoogleScholarGoogle Scholar | 16061880PubMed |
[129] R. D. Hancock, J. H. Reibenspies, H. Maumela, Inorg. Chem. 2004, 43, 2981.
| Crossref | GoogleScholarGoogle Scholar | 15106988PubMed |
[130] H. Maumela, R. D. Hancock, L. Carlton, J. H. Reibenspies, K. P. Wainwright, J. Am. Chem. Soc. 1995, 117, 6698.
| Crossref | GoogleScholarGoogle Scholar |
[131] K. E. Baidoo, D. E. Milenic, M. W. Brechbiel, Nucl. Med. Biol. 2013, 40, 592.
| Crossref | GoogleScholarGoogle Scholar | 23602604PubMed |
[132] J. L. Lange, P. R. W. J. Davey, M. T. Ma, J. M. White, A. Morgenstern, F. Bruchertseifer, P. J. Blower, B. M. Paterson, Dalton Trans. 2020, 49, 14962.
| Crossref | GoogleScholarGoogle Scholar | 33079111PubMed |
[133] B Bartoś, K Lyczko, A Kasperek, S Krajewski, A Bilewicz, J. Radioanal. Nucl. Chem. 2013, 295, 205.
| Crossref | GoogleScholarGoogle Scholar | 26224928PubMed |
[134] G. Montavon, A. Le Du, J. Champion, T. Rabung, A. Morgenstern, Dalton Trans. 2012, 8615.
| Crossref | GoogleScholarGoogle Scholar | 22678751PubMed |
[135] G. J. Beyer, M. Miederer, S. Vranješ-Đurić, J. J. Čomor, G. Künzi, O. Hartley, R. Senekowitsch-Schmidtke, D. Soloviev, F. Buchegger, ISOLDE Collaboration Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 547.
| Crossref | GoogleScholarGoogle Scholar | 14722680PubMed |
[136] C. Müller, J. Reber, S. Haller, H. Dorrer, U. Köster, K. Johnston, K. Zhernosekov, A. Türler, R. Schibli, Pharmaceuticals 2014, 7, 353.
| Crossref | GoogleScholarGoogle Scholar | 24633429PubMed |
[137] C. Vermeulen, G. F. Steyn, F. Szelecsényi, Z. Kovács, K. Suzuki, K. Nagatsu, T. Fukumura, A. Hohn, T. N. van der Walt, Nucl. Instrum. Methods 2012, 275, 24.
| Crossref | GoogleScholarGoogle Scholar |
[138] N. G. Zaitseva, S. N. Dmitriev, O. D. Maslov, L. G. Molokanova, G. Y. Starodub, S. V. Shishkin, T. V. Shishkina, G. J. Beyer, Czech. J. Phys. 2003, 53, A455.
| Crossref | GoogleScholarGoogle Scholar |
[139] R. E. Connick, J. Chem. Soc. 1949, S235.
| Crossref | GoogleScholarGoogle Scholar |
[140] M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche, W. J. Evans, J. Am. Chem. Soc. 2013, 135, 9857.
| Crossref | GoogleScholarGoogle Scholar | 23697603PubMed |
[141] C. T. Palumbo, I. Zivkovic, R. Scopelliti, M. Mazzanti, J. Am. Chem. Soc. 2019, 141, 9827.
| Crossref | GoogleScholarGoogle Scholar | 31194529PubMed |
[142] T. Kimura, Y. Kato, J. Alloys Compd. 1998, 278, 92.
| Crossref | GoogleScholarGoogle Scholar |
[143] T. I. Kostelnik, C. Orvig, Chem. Rev. 2019, 119, 902.
| Crossref | GoogleScholarGoogle Scholar | 30379537PubMed |
[144] C. Jiao, R. Zhong, Y. Zhou, H. Zhang, Int. J. Polym. Sci. 2020, 2020, 2175259.
| Crossref | GoogleScholarGoogle Scholar |
[145] R. Janicki, A. Mondry, Dalton Trans. 2019, 3380.
| Crossref | GoogleScholarGoogle Scholar | 30785145PubMed |
[146] M. Woods, S. Aime, M. Botta, J. A. K. Howard, J. M. Moloney, M. Navet, D. Parker, M. Port, O. Rousseaux, J. Am. Chem. Soc. 2000, 122, 9781.
| Crossref | GoogleScholarGoogle Scholar |
[147] C. A. Umbricht, U. Koster, P. Bernhardt, N. Gracheva, K. Johnston, R. Schibli, N. P. van der Meulen, C. Muller, Sci. Rep. 2019, 9, 17800.
| Crossref | GoogleScholarGoogle Scholar | 31780798PubMed |
[148] C. Müller, K. Zhernosekov, U. Köster, K. Johnston, H. Dorrer, A. Hohn, N. T. van der Walt, A. Türler, R. Schibli, J. Nucl. Med. 2012, 53, 1951.
| Crossref | GoogleScholarGoogle Scholar | 23139086PubMed |
[149] C. Muller, C. Vermeulen, U. Koster, K. Johnston, A. Turler, R. Schibli, N. P. van der Meulen, EJNMMI Radiopharm. Chem. 2017, 1, 5.
| Crossref | GoogleScholarGoogle Scholar | 29564382PubMed |
[150] P. Vojtíšek, P. Cígler, J. Kotek, J. Rudovský, P. Hermann, I. Lukeš, Inorg. Chem. 2005, 44, 5591.
| Crossref | GoogleScholarGoogle Scholar | 16060608PubMed |
[151] N. A. Thiele, J. J. Woods, J. J. Wilson, Inorg. Chem. 2019, 58, 10483.
| Crossref | GoogleScholarGoogle Scholar | 31246017PubMed |
[152] B. L. Garashchenko, V. A. Korsakova, R. Y. Yakovlev, Phys. At. Nucl. 2018, 81, 1515.
| Crossref | GoogleScholarGoogle Scholar |
[153] I. Murray, B. Rojas, J. Gear, R. Callister, A. Cleton, G. D. Flux, Cancer Biother. Radiopharm. 2020, 35, 530.
| Crossref | GoogleScholarGoogle Scholar | 32429699PubMed |
[154] F. Ortu, A. Formanuik, J. R. Innes, D. P. Mills, Dalton Trans. 2016, 7537.
| Crossref | GoogleScholarGoogle Scholar | 27094204PubMed |
[155] C. D. Tutson, A. E. V. Gorden, Coord. Chem. Rev. 2017, 333, 27.
| Crossref | GoogleScholarGoogle Scholar |
[156] F. Réal, M. Trumm, V. Vallet, B. Schimmelpfennig, M. Masella, J.-P. Flament, J. Phys. Chem. B 2010, 114, 15913.
| Crossref | GoogleScholarGoogle Scholar | 21070066PubMed |
[157] E. Bentouhami, G. M. Bouet, J. Meullemeestre, F. Vierling, M. A. Khan, C. R. Chim. 2004, 7, 537.
| Crossref | GoogleScholarGoogle Scholar |
[158] T. Ramdahl, H. T. Bonge-Hansen, O. B. Ryan, S. Larsen, G. Herstad, M. Sandberg, R. M. Bjerke, D. Grant, E. M. Brevik, A. S. Cuthbertson, Bioorg. Med. Chem. Lett. 2016, 26, 4318.
| Crossref | GoogleScholarGoogle Scholar | 27476138PubMed |
[159] K. Washiyama, R. Amano, J. Sasaki, S. Kinuya, N. Tonami, Y. Shiokawa, T. Mitsugashira, Nucl. Med. Biol. 2004, 31, 901.
| Crossref | GoogleScholarGoogle Scholar | 15464392PubMed |
[160] M. G. Ferrier, Y. Li, M.-K. Chyan, R. Wong, L. Li, S. Spreckelmeyer, D. K. Hamlin, T. Mastren, M. E. Fassbender, C. Orvig, D. S. Wilbur, J. Labelled Comp. Radiopharm. 2020, 63, 502.
| Crossref | GoogleScholarGoogle Scholar | 32812275PubMed |
[161] G. J. Deblonde, M. Sturzbecher-Hoehne, R. J. Abergel, Inorg. Chem. 2013, 52, 8805.
| Crossref | GoogleScholarGoogle Scholar | 23855806PubMed |
[162] T. A. Pham, J. Xu, K. N. Raymond, J. Am. Chem. Soc. 2014, 136, 9106.
| Crossref | GoogleScholarGoogle Scholar | 24870296PubMed |
[163] G. J. Deblonde, T. D. Lohrey, C. H. Booth, K. P. Carter, B. F. Parker, A. Larsen, R. Smeets, O. B. Ryan, A. S. Cuthbertson, R. J. Abergel, Inorg. Chem. 2018, 57, 14337.
| Crossref | GoogleScholarGoogle Scholar | 30372069PubMed |
[164] S. Hammer, U. B. Hagemann, S. Zitzmann-Kolbe, A. Larsen, C. Ellingsen, S. Geraudie, D. Grant, B. Indrevoll, R. Smeets, O. von Ahsen, A. Kristian, P. Lejeune, H. Hennekes, J. Karlsson, R. M. Bjerke, O. B. Ryan, A. S. Cuthbertson, D. Mumberg, Clin. Cancer Res. 2020, 26, 1985.
| Crossref | GoogleScholarGoogle Scholar | 31831560PubMed |
[165] N. Abbas, H. Heyerdahl, O. S. Bruland, J. Borrebæk, J. Nesland, J. Dahle, EJNMMI Res. 2011, 1, 18.
| Crossref | GoogleScholarGoogle Scholar | 22214432PubMed |
[166] I. Captain, G. J. P. Deblonde, P. B. Rupert, D. D. An, M.-C. Illy, E. Rostan, C. Y. Ralston, R. K. Strong, R. J. Abergel, Inorg. Chem. 2016, 55, 11930.
| Crossref | GoogleScholarGoogle Scholar | 27802058PubMed |
[167] G. T. Kent, G. Wu, T. W. Hayton, Inorg. Chem. 2019, 58, 8253.
| Crossref | GoogleScholarGoogle Scholar | 31198040PubMed |
[168] A. H. Staudacher, E. Bezak, A. Borysenko, M. P. Brown, Nucl. Med. Commun. 2014, 35, 1284.
| Crossref | GoogleScholarGoogle Scholar | 25192189PubMed |
[169] M. P. Kelley, G. J. P. Deblonde, J. Su, C. H. Booth, R. J. Abergel, E. R. Batista, P. Yang, Inorg. Chem. 2018, 57, 5352.
| Crossref | GoogleScholarGoogle Scholar | 29624372PubMed |
[170] P. E. Riley, K. Abu-Dari, K. N. Raymond, Inorg. Chem. 1983, 22, 3940.
| Crossref | GoogleScholarGoogle Scholar |
[171] U. B. Hagemann, C. Ellingsen, J. Schuhmacher, A. Kristian, A. Mobergslien, V. Cruciani, K. Wickstroem, C. A. Schatz, C. Kneip, S. Golfier, R. Smeets, S. Uran, H. Hennekes, J. Karlsson, R. M. Bjerke, O. B. Ryan, D. Mumberg, K. Ziegelbauer, A. S. Cuthbertson, Clin. Cancer Res. 2019, 25, 4723.
| Crossref | GoogleScholarGoogle Scholar | 31064781PubMed |
[172] K. Wickstroem, U. B. Hagemann, V. Cruciani, A. M. Wengner, A. Kristian, C. Ellingsen, G. Siemeister, R. M. Bjerke, J. Karlsson, O. B. Ryan, L. Linden, D. Mumberg, K. Ziegelbauer, A. S. Cuthbertson, J. Nucl. Med. 2019, 60, 1293.
| Crossref | GoogleScholarGoogle Scholar | 30850485PubMed |
[173] U. B. Hagemann, K. Wickstroem, E. Wang, A. O. Shea, K. Sponheim, J. Karlsson, R. M. Bjerke, O. B. Ryan, A. S. Cuthbertson, Mol. Cancer Ther. 2016, 15, 2422.
| Crossref | GoogleScholarGoogle Scholar | 27535972PubMed |
[174] A. V. Severin, A. N. Vasiliev, A. V. Gopin, I. E. Vlasova, E. V. Chernykh, Radiochemistry 2019, 61, 339.
| Crossref | GoogleScholarGoogle Scholar |
[175] W. Gaweda, M. Pruszynski, E. Cedrowska, M. Rodak, A. Majkowska-Pilip, D. Gawel, F. Bruchertseifer, A. Morgenstern, A. Bilewicz, Nanomaterials 2020, 10, 2067.
| Crossref | GoogleScholarGoogle Scholar |
[176] M. Gott, J. Steinbach, C. Mamat, Open Chem. 2016, 14, 118.
| Crossref | GoogleScholarGoogle Scholar |
[177] S. Hurst, Radium in Groundwater 2015 (Springer International Publishing: Cham).
[178] N. A. Thiele, S. N. MacMillan, J. J. Wilson, J. Am. Chem. Soc. 2018, 140, 17071.
| Crossref | GoogleScholarGoogle Scholar | 30485079PubMed |
[179] F. Reissig, D. Bauer, M. Ullrich, M. Kreller, J. Pietzsch, C. Mamat, K. Kopka, H. J. Pietzsch, M. Walther, Pharmaceuticals 2020, 13, 272.
| Crossref | GoogleScholarGoogle Scholar |
[180] D. S. Abou, N. A. Thiele, N. T. Gutsche, A. Villmer, H. Zhang, J. J. Woods, K. E. Baidoo, F. E. Escorcia, J. J. Wilson, D. L. J. Thorek, Chem. Sci. 2021, 12, 3733.
| Crossref | GoogleScholarGoogle Scholar | 34163647PubMed |
[181] M. Gott, P. Yang, U. Kortz, H. Stephan, H. J. Pietzsch, C. Mamat, Chem. Commun. 2019, 7631.
| Crossref | GoogleScholarGoogle Scholar |
[182] D. Bauer, M. Gott, J. Steinbach, C. Mamat, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 199, 50.
| Crossref | GoogleScholarGoogle Scholar | 29567522PubMed |
[183] D. Bauer, M. Blumberg, M. Köckerling, C. Mamat, RSC Adv. 2019, 9, 32357.
| Crossref | GoogleScholarGoogle Scholar |
[184] J. Steinberg, D. Bauer, F. Reissig, M. Kockerling, H. J. Pietzsch, C. Mamat, ChemistryOpen 2018, 7, 432.
| Crossref | GoogleScholarGoogle Scholar | 29928566PubMed |
[185] D. Abou, N. Thiele, A. Villmer, N. Gustche, F. Escorcia, J. Wilson, D. Thorek, J. Nucl. Med. 2020, 61, 587.
[186] E. Boros, J. P. Holland, J. Labelled Comp. Radiopharm. 2018, 61, 652.
| Crossref | GoogleScholarGoogle Scholar | 29230857PubMed |
[187] V. Frantellizzi, L. Cosma, G. Brunotti, A. Pani, A. Spanu, S. Nuvoli, F. D. Cristofaro, L. Civitelli, G. D. Vincentis, Cancer Biother. Radiopharm. 2020, 35, 437.
| Crossref | GoogleScholarGoogle Scholar | 31967907PubMed |
[188] J. Kozempel, O. Mokhodoeva, M. Vlk, Molecules 2018, 23, 581.
| Crossref | GoogleScholarGoogle Scholar |
[189] A. Piotrowska, S. Męczyńska-Wielgosz, A. Majkowska-Pilip, P. Koźmiński, G. Wójciuk, E. Cędrowska, F. Bruchertseifer, A. Morgenstern, M. Kruszewski, A. Bilewicz, Nucl. Med. Biol. 2017, 47, 10.
| Crossref | GoogleScholarGoogle Scholar | 28043005PubMed |
[190] M. Czerwinska, G. Fracasso, M. Pruszynski, A. Bilewicz, M. Kruszewski, A. Majkowska-Pilip, A. Lankoff, Materials 2020, 13, 3875.
| Crossref | GoogleScholarGoogle Scholar |
[191] F. Reissig, K. Zarschler, R. Hubner, H. J. Pietzsch, K. Kopka, C. Mamat, ChemistryOpen 2020, 9, 797.
| Crossref | GoogleScholarGoogle Scholar | 32775141PubMed |
[192] F. Reissig, R. Hübner, J. Steinbach, H.-J. Pietzsch, C. Mamat, Inorg. Chem. Front. 2019, 6, 1341.
| Crossref | GoogleScholarGoogle Scholar |
[193] A. N. Vasiliev, A. Severin, E. Lapshina, E. Chernykh, S. Ermolaev, S. Kalmykov, J. Radioanal. Nucl. Chem. 2017, 311, 1503.
| Crossref | GoogleScholarGoogle Scholar |
[194] P. Suchánková, E. Kukleva, K. Štamberg, P. Nykl, M. Vlk, J. Kozempel, RSC Adv. 2020, 10, 3659.
| Crossref | GoogleScholarGoogle Scholar |
[195] E.-A. Salvanou, D. Stellas, C. Tsoukalas, B. Mavroidi, M. Paravatou-Petsotas, N. Kalogeropoulos, S. Xanthopoulos, F. Denat, G. Laurent, R. Bazzi, S. Roux, P. Bouziotis, Pharmaceutics 2020, 12, 188.
| Crossref | GoogleScholarGoogle Scholar |
[196] R. M. Pallares, P. Agbo, X. Liu, D. D. An, S. S. Gauny, S. E. Zeltmann, A. M. Minor, R. J. Abergel, ACS Appl. Mater. Interfaces 2020, 12, 40078.
| Crossref | GoogleScholarGoogle Scholar | 32805833PubMed |
[197] E. Cędrowska, M. Pruszyński, W. Gawęda, M. Żuk, P. Krysiński, F. Bruchertseifer, A. Morgenstern, M.-A. Karageorgou, P. Bouziotis, A. Bilewicz, Molecules 2020, 25, 1025.
| Crossref | GoogleScholarGoogle Scholar |