

Imaging Cannabinoid Receptors: A Brief Collection of Covalent and Fluorescent Probes for CB1 and CB2 Receptors
Alexander J. Hamilton A , Alan D. Payne B , Mauro Mocerino B and Hendra Gunosewoyo A C
A C
A Curtin Medical School, Faculty of Health Sciences, Curtin University, Perth, WA 6102, Australia.
B School of Molecular and Life Sciences, Curtin University, Perth, WA 6102, Australia.
C Corresponding author. Email: Hendra.Gunosewoyo@curtin.edu.au

Mr Alex Hamilton is a Ph.D. student studying medicinal and organic chemistry at Curtin University. His research consists of designing novel cannabinoid receptor ligands and PARP-1 inhibitors from a versatile indole scaffold, with hopes of repurposing these compounds for further diseases and infections. |

Dr Alan Payne is a synthetic organic chemist at Curtin University. His main research interests are in designing new leads for drug discovery, antagonists of ethylene action in plants and fundamental hydrocarbons. Recently, he has focused on natural products from Western Australian plants as scaffolds for drug discovery. |

Professor Mauro Mocerino is a teaching and research academic at Curtin University with research interest in synthetic chemistry and chemistry education. The synthetic chemistry focuses on the design and synthesis of molecules for specific intermolecular interactions including chiral recognition and drug-protein interactions, the latter focusing on anti-protozoans or human papillomavirus inhibitors. The educational research is currently exploring the potential of virtual reality for visualisation of chemical processes. |

Dr Hendra Gunosewoyo is a synthetic medicinal chemist at Curtin University. His research is focused on the design and discovery of novel antimycobacterial molecules in collaboration with Johns Hopkins University. More recently, his research interests include medicinal chemistry of synthetic cannabimimetics and discovering other potential uses of indoleamides scaffold. |
Australian Journal of Chemistry 74(6) 416-432 https://doi.org/10.1071/CH21007
Submitted: 5 January 2021 Accepted: 13 April 2021 Published: 27 May 2021
Journal Compilation © CSIRO 2021 Open Access CC BY
Abstract
There has been an expanding public interest towards the notion that modulation of the sophisticated endocannabinoid system can lead to various therapeutic benefits that are yet to be fully explored. In recent years, the drug discovery paradigm in this field has been largely based on the development of selective CB2 receptor agonists, avoiding the unwanted CB1 receptor-mediated psychoactive side effects. Mechanistically, target engagement studies are crucial for confirming the ligand–receptor interaction and the subsequent biological cascades that lead to the observed therapeutic effects. Concurrently, imaging techniques for visualisation of cannabinoid receptors are increasingly reported in the literature. Small molecule imaging tools ranging from phytocannabinoids such as tetrahydrocannabinol (THC) and cannabidiol (CBD) to the endocannabinoids as well as the purely synthetic cannabimimetics, have been explored to date with varying degrees of success. This Review will cover currently known photoactivatable, electrophilic, and fluorescent ligands for both the CB1 and CB2 receptors. Structural insights from techniques such as ligand-assisted protein structure (LAPS) and the discovery of novel allosteric modulators are significant additions for better understanding of the endocannabinoid system. There has also been a plethora of fluorescent conjugates that have been assessed for their binding to cannabinoid receptors as well as their potential for cellular imaging. More recently, bifunctional probes containing either fluorophores or electrophilic tags are becoming more prevalent in the literature. Collectively, these molecular tools are invaluable in demonstrating target engagement within the human endocannabinoid system.
Keywords: imaging agents, cannabinoids, fluorescent probes, electrophilic probes, photoaffinity labelling, synthetic cannabinoids, cannabinoid receptor, ligand-assisted protein structure.
Introduction
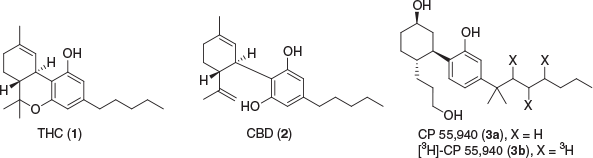
Cannabis sativa has been a widely used plant throughout civilisation for a multitude of different uses, namely as a recreational drug and for its medical purposes, as well as for industrial uses. Recent evidence has suggested Cannabis has beneficial therapeutic effects, which have been related to the active compounds located in the plant.[1–3] Several constituents known thus far in Cannabis, including the psychoactive (–)-trans-Δ9-tetrahydrocannabinol (THC or Δ9-THC) (1) and the non-euphoric cannabidiol (CBD) (2), were isolated and characterised in the late 1960s, are now termed cannabinoids (Fig. 1).[4,5] Subsequently, the human biological pathway that these cannabinoids interact with, was discovered and titled the endocannabinoid system (ECS).
|
The ECS consists of two known receptors, cannabinoid type 1 receptor (CB1R) and cannabinoid type 2 receptor (CB2R), the endogenous ligands (endocannabinoids), and the associated enzymes that break down and synthesise these ligands. CB1R was identified and characterised in rat brain in 1988 and verified by receptor localisation in 1990 using tritiated CP 55,940 (3a, 3b).[6,7] The receptor was cloned and the DNA that encodes the G protein-coupled receptor (GPCR) was found in the same year.[8] CB2R was discovered and cloned in 1993 and was primarily found to be localised in macrophages from the spleen.[9] The identification of these two important receptors that mediate the effects of Cannabis sativa constituents eventually resulted in the discovery of anandamide (AEA) (6) and 2-arachidonoyl glycerol (2-AG) (9) as the endogenous ligands that activate these receptors (Fig. 2).

|
The eicosanoids, AEA and 2-AG, were first identified and characterised in 1992 and 1995, respectively.[10–12] AEA is a partial agonist at both mammalian CB1R and CB2R (Ki(CB1R) = 61–543 nM; Ki(CB2R) = 279–1940 nM), whereas 2-AG is a full agonist at CB1R and a partial agonist at CB2R (Ki(CB1R) = 58.3–472 nM; Ki(CB2R) = 145–1400 nM).[13] The enzymes related to the biosynthesis of these endocannabinoids have also been identified. N-Acylation of phosphatidylethanolamine (4) via acyltransferase generates N-arachidonoyl-phosphatidylethanolamine (NAPE) (5), which is further catalysed by NAPE-hydrolysing phospholipase D (NAPE-PLD) to produce AEA and several other N-acylethanolamines.[14] Fatty acid amide hydrolase (FAAH) hydrolyses AEA into arachidonic acid (AA) (10), as well as various other endogenous amides and N-acylethanolamines.[15] On the other hand, 2-AG is synthesised from phosphatidylinositol 4,5-bisphosphate (PIP2) (7) to diacylglycerol (DAG) (8) via N-arachidonoyl phosphatidyl ethanolamine-preferring phospholipase D (PLCβ). DAG is then hydrolysed by diacylglycerol lipase α and β (DAGLα/β) to yield 2-AG, which is further hydrolysed by monoacylglycerol lipase (MAGL) to AA.[16] Similar to NAPE-PLD, DAGLα/β and MAGL are not exclusive to the 2-AG biopathway and are responsible for enzymatic catalysis of several other monoacylglycerols.[17,18] Inhibition of MAGL and FAAH has been shown to negate inflammation associated with neurological disorders, inducing analgesia, or are potentially a safer alternative to CB1R agonists.[19–21] However, due to the multitude of biological pathways these enzymes contribute to, their inhibition can also lead to undesirable and unpredictable side effects.[22]
In recent years, the crystal structures of CB1R and CB2R have been reported, complexed with ligands AM6538 and AM10257, respectively.[23–25] Two orphan receptors, GPR55 and GPR119, have been identified to be activated by classical cannabinoids such as CP 55,490 and various endogenous acylethanolamides.[26,27] The THC and AEA derivative, N-arachidonoyl-glycine, acts as a full agonist at the putative cannabinoid receptor GPR18, which is expressed in microglia suggesting a role in neuromodulation.[28–30] Transient receptor potential vanilloid 1 receptor (TRPV1) has also been shown to be activated by the two major endocannabinoids, AEA and 2-AG.[31] Interestingly, TRPV1 is co-localised alongside CB1R and has been shown to be modulated by non-psychotropic phytocannabinoids.[32] Due to the complexity of the ECS and the multitude of potential ligand–protein interactions, different screening assays can produce varying results, indicating probable off target activity or a differing mode of action.[33]
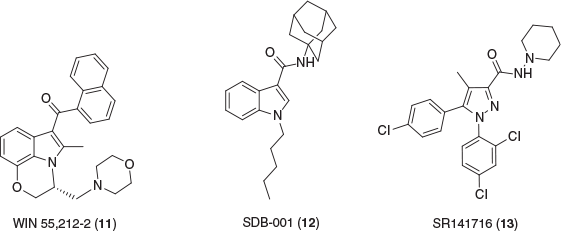
THC is the primary psychoactive component of Cannabis and a partial agonist at CB1R and CB2R, yet over 100 unique phytocannabinoids have been isolated, many of which inducing no euphoric side effects.[34] CB1R is primarily located in the central nervous system whereas CB2R is predominantly expressed in the peripheral immune system, however, both receptors are located vice versa in a lesser abundance.[35] The hippocampus, cerebellum, cerebral cortex, basal ganglia, and amygdala have been identified to be highly populated with CB1R.[36,37] Generally, CB1R agonists have negative side effects such as a reduction in motor coordination, memory, and cognition, thus limiting the therapeutic potential. WIN 55,212-2 (11) and SDB-001 (12) (Fig. 3) are two known potent full agonists at both cannabinoid receptors, producing similar effects to THC.[38–40] Rimonabant (SR141716) (13) is a CB1R inverse agonist used as a treatment for obesity but was withdrawn due to extreme side effects.[41–44] However, several phytocannabinoids have been shown to have a wide range of medicinal uses for treatment and protection of several neuroimmune disorders.[45–47] Notably, CBD is of interest due to its promising therapeutic potential—FDA approved as treatment for juvenile epilepsy—while containing no psychoactive properties.[48–50] Despite having low affinity to CB receptors, CBD has been shown to be a negative allosteric modulator at CB1R as well as being a partial agonist at CB2R.[51,52] While allosteric modulation of CB1R provides a promising therapeutic avenue, activating CB2R shows favourable treatment potential for a variety of neurological conditions.[53–55] CB2R is heavily expressed in microglia cells, prominently involved in several neuroinflammatory diseases such as Alzheimer’s, multiple sclerosis, and epilepsy. Blockage or inhibition of microglia has been shown to contribute to neuroprotection, by preventing production of proinflammatory mediators.[56] Several CB2R agonists, including phytocannabinoids, classical cannabinoids, and synthetic aminoalkylindoles, have displayed reductions in microglia activation in neurological diseases.[57–59]
|
The therapeutic potential of the endocannabinoid system and CB2R agonists have been well established, providing insights into potential selective drug design.[60,61] However, development of new drugs relies on understanding ligand–protein interactions or target engagement and the mechanism of action.[62] GPCRs are expressed in low levels in endogenous tissue, making target engagement difficult to identify, and is further accentuated by the adaptable nature of proteins when activated. Biomarkers are a useful tool for identifying target engagement in vivo by measuring the therapeutic effect, however this does not elucidate the compounds inability to interact with the receptor.[63] There are no biomarkers currently available for CB2R, increasing the demand for selective imaging methods and reducing off target interaction, although it is suggested changes in the ECS could be utilised as a biomarker for psychiatry.[33,64–66]
Small molecule probes serve as a comprehensive tool to image and identify information relating to ligand–receptor interactions and the applicability to disease treatment.[67] Positron emission tomography (PET) compounds have been widely utilised for verifying that the ligand binds with the target receptor via a radioisotope.[68] PET probes cannot only image the relevant proteins but are also capable of identifying biochemical processes at a molecular level before detectable anatomical variations. PET probes are utilised to determine progression or any alterations in a specific target’s activity. PET probes have been widely used for medicinal imaging in a multitude of fields such as oncology, cardiology, and neurology, with continued use and further research.[69–72] Some major drawbacks of PET probes include requiring on site synthesis, exposure to radiation, short lived radionuclides, and overall cost to run. PET probes also require a lower nanomolar affinity to be applicable for imaging purposes, although sometimes yielding false-positive results. Cannabinoid PET tracers have been comprehensively covered in several review articles.[73,74] Other predominant methods used for small molecule-target engagement are fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET) which are also used extensively.[75] Both BRET and FRET have been used to image several GPCRs, displaying the applicability of the methods in ligand–protein visualisation.[76,77] However, both methods require modification of the protein to include a fluorescent donor, which can ultimately alter the protein’s structure.[75] Several other imaging techniques have been applied to small cannabimimetic molecules. This review will focus on the known cannabinoid receptor type 1 and type 2 probes utilising non-invasive methods, namely electrophilic and photoactivatable probes, as well as fluorescent tagged ligands.
Photoactivatable and Electrophilic Probes
Small molecules that can covalently bind to specific amino acid side chains in certain receptors are a valuable tool in biological imaging. In GPCRs for example, the ligand first interacts with the target receptor through non-covalent binding, and ideally with high affinity. Once engaged with the target protein, the activatable group covalently binds to an amino acid residue under specific conditions. Electrophilic ligands are modified to contain an electrophilic group, e.g. an isothiocyanate (NCS) group, which reacts with a nucleophilic amino acid, generally lysine, histidine, and cysteine, to form the covalent bond in situ without significant alteration of biological activity. Photoactivatable (or photoaffinity) based ligands have an inert group, generally azides, benzophenones, or diazirine moieties, which upon irradiation form a reactive species that covalently binds with nearby amino acids irreversibly.[78,79] Photoactivatable ligands have an advantage in comparison to electrophilic ligands due to the in situ reactive species of the latter binding to any nearby amino acids.[80] Monofunctional ligands containing one group have been primarily applied to receptor binding studies, however bifunctional ligands are becoming of more interest. Once the compound has irreversibly attached to the target protein, the difference in Bmax via a radioligand binding assay determines this degree of covalent interaction. Despite the advantageous use of studying ligand–protein interactions, a major drawback for the ligands is the lack of imaging potential. This can be circumvented by the addition of a second reactive group, utilising click chemistry to attach a photoprobe in situ, giving the imaging potential.[79,81] Since the early 1990s, several electrophilic and photoactivatable cannabinoids have been synthesised (Fig. 4, Table 1), some of which assist in determining crucial amino acid residues in ligand–receptor binding.

|

|
Classical Cannabinoids
Several covalent cannabinoids containing similar scaffolds to endo and phytocannabinoids were initially synthesised in the early 1990s. A photoactivatable ligand, 5′azido-Δ8-THC or AM91 (14), showed a 2-fold increase in binding affinity when compared with its phytocannabinoid counterpart, (–)-Δ8-THC (Ki(rCB1R) = 35 ± 11 nM) against [3H]-CP 55,940.[82] This ligand demonstrated the potential of photolabelling by labelling the receptor active site and the group would intend to develop further probes for identification of cannabinoid receptor subtypes. The radioiodinated equivalent, (–)-2-iodo-5′azido-Δ8-THC (15), when exposed to UV light was found to irreversibly bind to sites in both a mouse cerebral cortex (Kd = 5.60 pM) and S49 mouse lymphoma cells (Kd = 9.38 pM).[83] The Makriyannis group noted due to several bands obtained from the SDS–PAGE and autoradiography, that multiple cannabinoid receptors might be present in heterogenous cells, which was confirmed later.
The same group further explored this scaffold, utilising isothiocyanate for covalent binding, to yield (–)-11-OH-7′-NCS-1,1′-dimethylheptyl-Δ8-THC (AM708) (16) as the first electrophilic cannabinoid ligand.[84] Pre-equilibration of rat forebrain membranes at 10 μM with this compound resulted in an 80 % decrease in [3H]-CP 55,940 binding at rCB1R and a 100 % decrease at 100 μM, with a nanomolar half maximal inhibitory concentration (IC50) value. An azido group at the terminus of the sidechain yielded AM839 (17), exhibiting potent binding, however no further studies have been conducted with this compound.[85] The results demonstrated that classical cannabinoids can be modified at the tail of the alkyl chain with electrophilic groups to irreversibly bind to the active site of the receptor. With this new insight, the Makriyannis group synthesised 7′-NCS-DMH-Δ8-THC (18) removing the hydroxy at position 11.[86] The IC50 value was comparable to the alkyl hydroxy derivative and similar [3H]-CP 55,940 displacement at 83 %. The probe showed a 3-fold higher affinity than AM708, indicating that the aliphatic hydroxy does not affect affinity.
Several variations of THC have been synthesised and published incorporating an iodine into the scaffold. The aliphatic azido group was altered to be positioned at C-9 of the tricyclic ring for two novel photoaffinity probes, AM869 (19) and AM1708 (20).[87] Both compounds were shown to have high affinity to CB1R and CB2R, with AM1708 being radioiodinated at the terminal end of the aliphatic chain as well as an olefin. In 2003, an iodo and isothiocyanate probe AM960 (21) with structural similarity to Nabilone was synthesised by Chu et. al.[88] Although very limited studies had been performed on the probe, it displayed successful irreversible covalent binding to rCB1R by occupying 50 % of sites at 25 nM. The radioiodinated equivalent however has not been synthesised or tested.
AM841 (22) further explored the scaffold of THC with the continued trend of altering the terminal end of the DMH (dimethylheptane) chain with NCS for covalent binding.[89] The compound exhibited high affinity to CB1R expressing CHO-K1 WT cells and displaced [3H]-CP 55,940 (Kd = 6.7 ± 0.34 nM) and [3H]-WIN 55,212–2 (Kd = 18.3 ± 0.99 nM). C6.47(355) is a cysteine residue located in transmembrane helix (TMH) 6 of CB1R that is of interest for ligand–receptor interaction. This study tested the binding potential of AM841 when the amino acid was mutated to less nucleophilic groups, serine, alanine, and leucine. Serine and alanine mutations showed little loss of affinity, whereas AM841 suffered a substantial loss of affinity with the mutated leucine residue. [3H]-CP 55,940 demonstrated a loss of affinity resulting from this mutation, while [3H]-WIN 55,212-2 remained unchanged. This data suggests that C6.47(355) plays a role in recognition of THC derived analogues but is not crucial for (aminoalkyl)indole compounds, indicating different binding motifs. This was supported by a reduction in [3H]-CP 55,940 binding to AM841 WT cells incubated for 1 h by 6-fold but was unobserved with [3H]-WIN 55,212-2.
Ligand-assisted protein structure (LAPS) is an analysis method in which high affinity probes covalently bind to amino acids in proximity to the target protein.[105] Specific amino acids are then mutated, to govern the effect of the binding ability of the probes with these alterations, ultimately determining important residues in binding. Once achieved, in silico molecular modelling of the protein–ligand interaction is developed to further validate the experimental findings. Identification of the binding site can then be supported by liquid chromatography and/or mass spectrometry proteomics via an attachment of the covalent ligand to the protein. Supported by the LAPS approach, a cysteine residue in TMH6, C6.47(257), was found to be an essential amino acid in CB2R binding with AM841.[90] It was revealed that C6.47(257) of hCB2R is homologous to C6.47(355) of hCB1R as per covalent binding of AM841. Altering C6.47(257) to serine and alanine in HEK-293 WT-hCB2R cells found similar displacement of [3H]-CP 55,940 and a 2-fold increase, respectively. The serine and alanine mutated receptors also found AM841 displaced [3H]-WIN 55,212-2 with a 3 and 6-fold greater affinity, respectively. AM841 binds covalently to the WT hCB2R, reducing the Bmax of [3H]-CP 55,940 by 80 %, whereas the C6.47(257)A and C6.47(257)S displayed no sign of irreversible binding. Mutation of two close proximity cysteine residues, C7.38(284) and C7.42(288), to serine did not affect the affinity of AM841 and hence it was concluded that C6.47(257) is the residue in which covalent attachment occurs. Interestingly, mutation of K3.28[109] in hCB2R resulted in a similar Ki to the WT and no saturable binding was noted via mutation of K3.28(192) in hCB1R, indicating hCB1R and hCB2R have unique binding regions.
Validation of C6.47(257) and the covalent interaction between AM841 was supported by multiple reaction mass spectrometry monitoring (MRM-MS).[106] The seven TMHs of hCB2R were first separated and exposed which confirmed AM841 binding to TMH6. Then, a competitive binding assay against [3H]-CP 55,940 in hCB2R overexpressed in Spodoptera frugiperda (Sf-21) cells as N-terminal FLAG-tagged/C-terminal 6His-tagged (FLAG-hCB2R-6His) yielded a similar Bmax decrease at ~75 % upon prior incubation with AM841. MRM-MS allowed visualisation of two discrete TMH6 peaks and incubation of AM841 in purified FLAG-hCB2R-6His displayed an ~75 % decrease of the TMH6 peak relative to that without AM841. Further high-resolution MS demonstrated that AM841 is selective to covalently modifying hCB2R at C6.47(257). AM841 has also been shown to have anti-inflammatory properties in central and peripheral mCB1R and mCB2R, in an in vitro and in vivo study of mouse models of colitis.[107] Another study demonstrated that AM841 is a peripheral restricted ligand, slowing GI motility in normal mice conditions and normalising accelerated motility in stressed mice.[108] Further studies of covalent ligands as a potential treatment of disease is an interesting route, however consideration should be taken due to the reactive and irreversible nature of the probes.
AM4703 (23) was first reported in 2017 as a derivative of AM841.[91] The NCS group is relocated to C-11, as opposed to the terminal end of the alkyl chain, to further expand upon cannabinoid binding. AM4703 displayed a high affinity to WT hCB2R HEK-293 cells against [3H]-CP 55,940 and was observed to be a potent irreversible agonist, although weaker than AM841. Covalent binding was found to be ~60 % at 33 nM in which similar results were obtained to mutated cysteine residues in TMH1, 6, and 7. However, [3H]-CP 55,940 specifically bound to C2.59(89)S was completely displaced indicating covalent interaction with this residue in TMH2 and a potent binding domain for classical cannabinoids. In confirmation with experimental data, molecular modelling of AM4703 with hCB2R suggests that classical orthosteric agonists bind to TMH2 and 3 as guided by the location of NCS. The study is the first to involve C2.59[89] in hCB2R ligand–receptor binding, providing new insights into classical cannabinoid motifs and binding regions.
Previous studies had shown that an arylphenone at C-3 of Δ8-THC displayed high affinity to CB2R. Focusing on modifying the C-3 alkyl chain to different arylphenone substituents, the Makriyannis group found that 3-benzothiophenyl (AM967) (24) had moderate affinity to mCB2R and hCB2R.[92] The photolabelling of this compound was performed on HEK-293 cells expressing mCB2R, exhibiting a 67 % photolabel to the receptor. The study revealed that 3-ketoaryl compounds have moderate selectivity to CB2R in mouse models and have the potential to photolabel in relatively high yields. Continuing this modification of the C-3, adamantyl groups with varying photoactivatable and electrophilic functionality at tertiary carbons where synthesised and tested. The two most potent compounds, AM993 (25) and AM994 (26), differed by an isothiocyanato and azide group through a methylene, respectively.[93] AM994 showed high affinity to CB1R with 3-fold and 10-fold selectivity over human and mouse CB2R, respectively. Pretreatment of the cells with AM994 reduced [3H]-CP 55,940 ability to bind by 63 and 74 % at CB1R and CB2R with 10-fold the compound’s Ki value. Similar to AM994, AM993 showed high affinity to CB1R with 2-fold and 6-fold selectivity over human and mouse CB2R. Covalent labelling via irradiation of the compound at 10-fold the Ki, yielded a reduction in [3H]-CP 55,940 binding by 67 % at CB1R and 60 % at CB2R. AM993 was tested before irradiation for covalent binding, observing no reduction in specific binding of [3H]-CP 55,940. Both AM994 and AM993 were found to be agonists at CB1R but showed negligible response at CB2R. These compounds were suggested to be used to obtain information about the structure of CB1R and were being explored for X-ray crystallography to determine ligand–receptor complexes.
Arylpyrazole Cannabinoids
With C6.47(355) being an essential acceptor for receptor–ligand labelling in CB1R, a selective covalent ligand, AM1336 (27), was designed to target cysteine residues in hCB2R.[94] Modifying the biarylpyrazole CB2R antagonist, SR144528, the first CB2R selective electrophilic probe, AM1336 was synthesised. AM1336 was found to bind irreversibly to hCB2R as 60 % of [3H]-CP 55,940 could not interact with the occupied receptor after pre-incubation and washing. AM1336 displayed a noticeable drop in covalent labelling to the mutated TMH7 residues, C7.38(284) and C7.42(288), indicating these amino acids are important for inverse agonist interactions. The binding competency of hCB2R overexpressed in Sf-21 cells (FlaghCB2his6) was tested by labelling with AM1336.[109] AM1336 (50 nM) showed 73 % labelling of the receptor, decreasing the Bmax of [3H]-CP 55,940. Mass spectrometry analysis of hCB2R complexed with AM3661 again identified C7.38(284) of TMH7 as important for inverse agonist binding in LAPS analysis. C6.47(257) was also noted to be of importance in the same study for agonist binding using AM841.
Indole Cannabinoids
In 1996, a range of electrophilic (aminoalkyl)indole compounds were synthesised based on the agonist Pravadoline.[95] From the compounds, isothiocyanate ‘12’ (28) was the most potent with a half maximal effective concentration (EC50) of 1.1 μM, with most other compounds being inactive at < 10 μM. rCB1R membranes incubated with 1 μM of isothiocyanate ‘12’ reduced [3H]-CP 55,940 binding by 70 %, demonstrating irreversible binding. At 100 μM, no further loss of [3H]-CP 55,940 binding was observed. Allosteric modulation of GPCRs can regulate pharmacological responses by altering the receptor conformation such that orthosteric ligand efficacy is modified.[110,111] The effect of an allosteric ligand binding can either give positive or negative responses, known as positive allosteric modulators (PAM) or negative allosteric modulators (NAM), respectively. Expanding on information about controlling the undesirable psychoactive effects of CB1R, allosteric modulation can help utilise the therapeutic potential of the ECS. Org27569 is a NAM with a moderate potency at CB1R (pEC50 = 8.64 ± 0.11 at 100 nM).[112] Looking for cysteine residues assisted by LAPS, GAT100 (29) was found to be the most effective of four compounds based off Org27569. The electrophilic ligand contains an isothiocyanate at C-5 of the indole, demonstrating higher potency than Org27569, and did not possess any inverse agonist activity.[96] Preincubation of HEK-293 cells with GAT100 at 100 nM increased the specific binding of [3H]-CP 55,940 at CB1R by over 2-fold, exhibiting covalent binding and negative allosteric modulation, whereas Org27569 had no effect. Ligand-docking and in silico analysis of GAT100 in complex with hCB1R determined C7.32(382) as the residue with which GAT100 feasibly covalently interacts.[97] The same study also found that GAT100 is an effective NAM in conjunction with AEA and 2-AG when evaluating its orthosteric-probe dependence. Electrophilic NAM cannabinoids could potentially elucidate new binding pockets in which allosteric modulation occurs, which could theoretically help future modulation of negative side effects of CB1R activation.
Endocannabinoids
Methyl arachidonyl fluorophosphonate (MAFP) (30) was found to be an irreversible CB1R antagonist reducing binding of classic cannabinoid agonists, WIN 55,212-2 and CP 55,940, while not affecting the binding abilities of non-cannabinoids. MAFP was also found to generate irreversible inhibition of phospholipase A2 and FAAH.[98,113] The Makriyannis group designed and characterised two high affinity AEA probes, AM3677 (31) and AM3661 (32), containing the electrophilic thioisocyanate and photoactive N3 at the terminal C-20 alkyl chain, respectively. Both compounds when compared with ACPA (Ki(rCB1R) = 2.7 ± 0.4 nM; Ki(rCB2R) = 157 ± 37 nM), showed an increase in affinity to both CB receptors. [3H]-CP 55,940 binding was lowered by 58 % in rat forebrain membranes when equilibrated with 26 nM of AM3677. The covalent binding was determined to reach maximum concentration as uptake plateaued at 30 min.[99] AM3677 was also found to inhibit forskolin-activated cAMP formation with an IC50 of 27.1 ± 2.2 nM in Flp-In-293 cells overexpressing CB1R.[100] One-hour preincubation of AM3677 displayed a 43 % irreversible binding to hCB1R, where [3H]-CP 55,940 was unable to interact with the same receptor. The same study found AM3677 binds covalently to hCB1R at C6.47(355), in which AM841 binds covalently as well, indicating this residue is critical to CB1R binding.[100] AM3661 was equilibrated at 3-fold the Ki, then irradiated with UV light (254 nm) which resulted in 23 % irreversible labelling of rCB1R.[99] When pre-incubated with AM3661, a reduction of 68 % to [3H]-CP 55,940 binding was exhibited at 18 nM. The results indicated that AM3661 could be a useful probe for rCB1R imaging, in elucidating the binding pocket, helping assist with future probe design. With evidence suggesting AEA novel receptors exist, Balas et al. produced a photoactivatable probe derived from endocannabinoids (33). The compound featured a 2-azido-5-iodobenzoate ester group, due to the advantage of wavelength activation higher than protein UV absorbance. The compound showed some affinity to both receptors in recombinant human receptors, with a decrease of affinity to hCB1R when compared with AEA. The compound was suggested to be a tool for discovery of potential new endocannabinoid receptors.[101] Balas et al. further synthesised AEA probes, based on the preliminary data. A replacement of the arylazide to 2-tert-butyl-2-methyl-1,3-benzodioxole-4-carboxylate (TBMB), removed all notable affinity to both receptors (Ki(hCB1R) = 2.80 ± 0.05 μM; Ki(hCB2R) = 1.02 ± 0.03 μM).[114] AM9017 (34) is an AEA derived covalent probe with the ability to potentially perform click reactions in situ.[102]
Bifunctional Cannabinoids
Bifunctional probes bare the ability to utilise a multitude of different imaging techniques to create a higher spatial resolution. A novel Δ8-THC analogue 35 utilises a diazirine as the photoactive handle and an alkyne for in situ click chemistry via the alkyl chain at C-3.[103] The compound displaced [3H]-CP 55,940 in CB1R and CB2R in overexpressing CHO cells with a high affinity to both receptors. The probe using a two-step photoaffinity labelling assay did not label either CB receptor at 2 μM, which suggested that changing the position of the photoreactive group to the rigid tricyclic core might permit covalent interaction with the receptor. The same group modified LEI101 to contain a terminal alkyne and a trifluoromethyl-diazirine-benzoyl, yielding LEI121 (36) as a CB2R photoaffinity compound.[104] The presence of the alkyne couples the fluorophore(s) by a click reaction to give the resulting triazole linked probe. The ligand without any linked probes was tested via a radio ligand binding competition assay using [3H]-CP 55,940 in hCB2R-expressing CHO cells. LEI101 (pKi(CB2R) < 7.5 ± 0.1) had a similar affinity to the novel probe, LEI121 (pKi(CB2R) < 7.2 ± 0.4), which underwent a slight loss of CB2R affinity while showing little CB1R affinity. After UV-irradiation of LEI121 bound to CB2R, specific binding of [3H]-CP 55,940 was substantially lowered, while the binding remained unchanged without light activation, indicating covalent binding to the protein. While LEI101 is reported to be a partial agonist, LEI121 was found to be an inverse agonist through two functional assays.
AM859 (37) is a unique compound that acts similarly to AM869, except functioning as a bifunctional covalent probe due to the terminal azide as opposed to an iodine. The probe displayed excellent affinity to both receptors with the hope of elucidating more information about ligand and receptor interaction. Several unpublished probes have been synthesised and reported by the Makriyannis group.[102] These probes are notable due to the bifunctional ability, either homo or hetero covalent ligands. AM5822 (38) and AM5823 (39) are both bifunctional probes with altering functional groups at the terminal end of the DMH chain. AM5823 offers the ability to covalently bind twice due to multiple isothiocyanate groups, whereas AM5822 contains an azide group for light activation. Although most probes are preliminary, the potential of bifunctional ligands to image the receptor at higher spatial resolution is becoming of more interest.
In the same study as AM4073, the bifunctional ligand AM4099 (40) contains an isothiocyanate at the terminus of the C-3 sidechain and at C-11 of the cyclic scaffold.[91] Interestingly, the probe does not bind to hCB2R C6.47(257) as AM841 does, instead binding to C2.59[89] like AM4073. Due to the similar binding affinities of AM4073 and AM4099, it is postulated that C6.47(257) orients AM841 within the hCB2R binding pocket to obtain ‘megagonist’ potency. This is heavily supported by the NCS lacking AM841 analogue, AM4056, yielding similar potency to AM4073 and AM4099.[90] While it is evident that the position of the electrophilic and photoactivatable group affects the binding and potency of the compound, further development of classical, endo, and synthetic cannabinoid motifs can be explored and aided by techniques, such as LAPS, for a more comprehensive understanding of ligand–protein binding.
Fluorescent Probes
Small molecules that contain a fluorescent moiety have been widely developed and applied to biological imaging. These compounds offer a safer and more accessible method of imaging when compared with radioligands, as they can be applied to in vivo studies in native systems and even act as a competing ligand in binding assays.[115,116] The difficulty of selective non-peptide fluorophore development has been well established, and a direct addition of a fluorescent scaffold can detrimentally affect affinity.[117] However, these compounds are designed with a linker, generally polyethylene glycol (PEG) or methylene chain, to ensure affinity of the parent compound remains.[118] Tissue penetration is another issue with fluorescent ligands; however, near-infrared (NIR) dyes can be imaged, minimising problematic auto-fluorescence within the biological system.[119,120] These fluorescent moieties must absorb and emit above ~600 nm, as well as having good chemical and photostability, for the dye to be applicable for receptor imaging. Quantum yields in aqueous and biological systems must also be considered when fluorescent probes are to be used for in vivo imaging, due to probable issues with detection. Fluorescent probes also show potential capabilities of direct examination of allosteric and orthosteric ligand–receptor interactions.[121] Within recent years, several known potent cannabinoids have been modified to contain fluorescent compounds (Fig. 5, Table 2), with the hopes of elucidating cannabinoid pharmacology.

|

|
Biotinylated Cannabinoids
Biotinylation is the addition of biotin to a ligand via a linker. Once the compound has docked with the target receptor, fluorescent avidin conjugates are used due to the specificity to interact with biotin, subsequently providing fluorescence to the protein.[143] Endogenous biotin can be blocked by an avidin conjugate to view wild-type and endogenous systems without undesirable interaction. A biotinylated equivalent of AEA (linked via a PEG chain), had similar lipophilicity to the endogenous ligand and was used to show the distribution of AEA, without interfering or interacting with other endogenous systems.[144] In 2011, several endocannabinoids were modified via biotinylation or an alkyne addition for in situ click reactions.[122] The structures synthesised structurally resemble AEA, 2-AG, and 2-arachidonyl glyceryl ether (2-AGE) in which all compounds are partial agonists. The most potent of the endocannabinoid derivatives belonged to 2-AG and 2-AGE, whereas AEA and methanandamide compounds showed little affinity. The potent 2-AG and 2-AGE derivatives were screened against [3H]-CP 55,940 in human CB receptor transfected HEK-293-EBNA cells. Both compounds showed an increase in CB2R affinity over 2-fold, and a decrease towards CB1R over 8-fold. The biotinylated 2-AG analogue was described to be a selective CB2R probe having little CB1R affinity (Ki(CB1R) > 5000 nM; Ki(CB2R) = 379 nM), whereas 2-AGE-3b (41) had retained moderate affinity to both receptors. 2-AGE-3b was selected for in vitro imaging of CB1R due to its affinity and was incubated in CB1R transfected mouse hippocampal cell line HT-22. Streptavidin-Alexa488 was used successfully to image the probe, visualising the receptor. To verify CB1R binding, the compound was tested in non-CB1R transfected cells against an excess of HU-210, to show little staining with the streptavidin. Non-specific binding was dismissed by co-labelling the compound with an anti-CB1 antibody and anti-rabbit Alexa633, in which both fluorescent compounds overlapped.
The research group continued by modifying potent synthetic cannabinoids with the biotin moiety.[123] HU-210 is a non-selective potent agonist at both CB1R and CB2R originally synthesised in 1988 by Mechoulam, mimicking the effects and structure of THC (Ki(CB1R) = 0.061 nM; Ki(CB2R) = 0.52 nM).[145] The Mechoulam group further explored this model and synthesised HU-308 in 1999, with more selectivity towards CB2R (Ki(CB1R) > 10000 nM; Ki(CB2R) = 22.7 nM).[146] The compounds synthesised were conjugated via the free hydroxy, either at C-1 or C-11, to biotin through a linker, in efforts for higher affinity probes.[123] These compounds were screened against [3H]-CP 55,940 in human CB receptor transfected HEK-293-EBNA cells. Out of the two biotinylated HU-210 derivatives, HU210-1 (42), had the highest affinity to both receptors with the C-1 biotin linked compound exhibiting an equally high affinity (Ki(CB1R) = 11 ± 2 nM; Ki(CB2R) = 3 ± 1 nM). The HU-308 linked with biotin, HU308-3 (43) maintained its selectivity to CB2R while only enduring a minor loss of affinity. Probes HU210-1 and HU308-3 were selected for in vitro imaging in native systems, labelling neurons in CB1R and microglia in CB2R, respectively.[123] Compound 42 labelled neurons in CB1R, detected by streptavidin-AlexaFluor488, and when co-labelled with anti-microtubule-associated protein 2 antibody plus AlexaFluor594, fluorescent overlapping was observed. Both 42 and 43 were successfully visualised at CB2R in rat microglial cells detected by streptavidin-AlexaFluor488. Additionally, an excess of HU-210 with either compound gave no fluorescent signal. The results indicated the first suitable probes for imaging both CB receptors with moderate affinities and specific binding.
Fluorescent Classical Cannabinoid Derivatives
The major drawbacks of a biotin probe, being two-step labelling and blocking endogenous biotin, results in the biotinylated cannabinoids not being suitable for tissue staining. Recently, AlexaFluor 488, with a hexyl linker was selected as the probe for HU-210 at the allylic hydroxyl (44).[124] A competitive radio ligand binding assay with [3H]-CP 55,940 using human CB1 and CB2 transfected HEK-293-EBNA cell membranes gave CB1R selectivity with moderate CB2R affinity (Ki(CB1R) = 27 ± 4 nM; Ki(CB2R) = 0.8 ± 0.2 μM). The fluorophore showed in vivo visualisation of human tonsil CB1-expressing cells using confocal microscopy. When compared with the non-selective binding properties of HU-210, the CB1 selectivity was obtained upon the addition of the AlexaFluor488 linker. This insight could lead to selective CB1R and CB2R fluorescent probes derived exclusively from HU-210.
Chromenopyrazole compounds, containing a similar structural design as THC and HU compounds, have been shown to have high affinity to cannabinoid receptors.[147] It was reported that phenolic alkylation of the pyrazole ring resulted in higher CB2R affinity, making this suitable for a linker position.[148] Coupled with three different fluorophores, BODIPY-630/650, BODIPY-FL, and Cy5, the phenolic chromenopyrazole scaffold was utilised for potential CB fluorescent ligands.[125] The C-4 position of the phenyl was found to have better linker tolerance and higher affinity to CB2R. Using BODIPY-630/650 as the fluorophore, the binding affinity (pKi(CB2R) = 5.80 ± 0.12) was deemed too low to be useful for imaging studies. Extending the PEG linker from 2 to 5 showed no improvement to the fluorophore’s imaging potential or selectivity for BODIPY-630/650. The BODIPY-FL attached linker exhibited an increase by ~10 fold to CB2R selectivity (pKi(CB2R) = 6.84 ± 0.04), suggesting a smaller fluorophore is more ideal. The highest CB2R affinity however came from the commercially available Cy5 dye with little CB1R interaction, showing promising potential for the scaffold 45 (pKi(CB1R) = 5.22 ± 0.11; pKi(CB2R) = 7.38 ± 0.05). The Cy5 compound was analysed via a cAMP BRET assay, revealing the compound to act as an inverse agonist, with higher potency than SR144528. Incubation of CB2R expressing HEK-293 cells with the Cy5 fluorophore demonstrated specific binding from a decrease in fluorescence when SR144528 is competing. Due to the potential issues of ligand with cell permeability, the probe is suggested to be beneficial for in vitro and ex vivo imaging, but no testing has been published thus far.
Fluorescent Indole Derivatives
JWH-015 is a potent cannabinoid agonist, containing a naphthoylindole scaffold, with roughly 28-fold the affinity to CB2R over CB1R. Due to the high selectivity of compounds at the time, one of the first fluorophores for CB imaging was designed and synthesised to contain the fluorescent dye, nitrobenzofurazan (NBD).[126] The NBD fluorophore 46 was linked through the naphthalene via a short amide link to prevent a non-fluorescent, PET active compound being produced.[149] CB2R affinity was determined by a displacement assay against [3H]-CP 55,940 in CHO cells expressing CB2R. The new NBD JWH-015 resulted in only a 25 % displacement at 10 μM. When visualised under a confocal microscope, most of the fluorescence was localised in the cytoplasm due to the lipophilicity of the compound and hence rapid cellular uptake. The precursors of the compound were found to still contain CB2R activity, however this avenue was not explored and in recent years indole compounds were found to primarily tolerate fluorophores at C-7 despite being antagonists.[150]
With this loss of affinity, employment of a potent CB2R antagonist scaffold, 6-methoxy-N-pentyl isatin acylhydrazone, was conjugated with the NBD dye.[127] NMP6 (47) was synthesised over two steps, utilising NBD to bind to the large hydrophobic pocket for potential CB2R affinity. NMP6 showed a Ki value of 387 nM in Chinese hamster ovary (CHO-K1) cells, when screened against [3H]-CP 55,940 in a competitive binding assay. The excitation and emission of NMP6 in acetonitrile were 470 and ~530 nm, respectively. Visualisation of the ligand was achieved by staining mouse T-cells with PE-conjugated anti-CD4 antibody, in which 5 μM of NMP6 was required to stain CD4+ T-cells. Pre-incubating the cells with GW842166X, a CB2R agonist, shows a decrease in cell staining due to a reduction in binding. Despite the scaffold showing reliable CB2R affinity, further analogues containing the isatin scaffold have not been tested as an avenue for CB imaging.
Fluorescent Pyrazole Derivatives
SR144528 was one of the first synthetic cannabinoids to act as an inverse agonist selectively at CB2R (Ki(hCB1R) = 400 nM; Ki(hCB2R) = 0.6 nM).[151] Despite having a high affinity and selectivity to CB2R, SR144528 was never used as an imaging agent, due to a lack of a conjugatable group. In 2008, mbc94 (48) was first synthesised with a benzyl bromide group in place of a methyl for coupling capabilities.[152] The near-infrared (NIR) dye, IRDye 800CW NHS ester, was coupled to mbc94 to give NIRmbc94 (49), the first reported CB2R selective probe. The probe itself has promising photophysical properties with a maximum absorbance of 779 nm and an emission of 797 nm in water. NIRmbc94 labelled CB2-expressing DBT cells from a mouse astrocytoma cell line, whereas the NIR dye incubated in the cells showed no signal. The fluorescent signal was reduced when non-CB2R expressing cells where incubated with the same concentration.
The mbc94 scaffold was further tested to investigate its affinity to CB2R. In 2011, a competitive binding assay against [3H]-CP 55,940 showed NIRmbc94 having a Ki of 260 nM in CB2 mid DBT cells, whereas the non-conjugated mbc94 had a Ki of 15 nM.[128] The same study found that NIRmbc94 binds specifically to CB2-mid DBT cells and does not bind to DBT cells without endogenous CB2R. These results allowed further competition curves against potent CB2R compounds, SR144528 and WIN 55,212-2. Both compounds competing against NIRmbc94 for CB2R binding gave Ki values of 4.7 and 3 nM, respectively. The NIRmbc94 signal was found to remain undisturbed with non-CB2R active compounds present and active in BV-2 cells with a Ki of 3 nM against WIN 55,212-2, showing binding to endogenous CB2R expressed by the mouse microglia cells.
Further expanding on NIRmbc94, the same group synthesised NIR760-mbc94 (50), with the same pharmacophore but a different fluorophore.[129] The new fluorophore, NIR760, has similar properties to IRDye 800CW with a different linker position to the pharmacophore. The absorbance for the new fluorescent probe is 766 nm and a fluorescence emission of 785 nm with a quantum yield of 15.2 %. In a saturated binding assay with SR144528, the NIR760-mbc94 binds to the CB2R with a Kd of 26.9 nM. NIR760-mbc94 incubated in CB2-mid DBT cells exhibited a 4-fold higher florescent signal than that of cells incubated with NIR760. In the presence of SR144528, the NIR760-mbc94 incubated cells showed 40 % lower fluorescence intensity. In vivo testing found NIR760-mbc94 increased the tumour-to-normal ratio by 3.7-fold at 72 h post-injection. The selective binding to CB2R, both in vitro and in vivo, suggests NIR760-mbc94 to be a promising imaging agent for CB2R-positive cancers.
With new zwitterionic NIR fluorescent probes being used for imaging, the mbc94 pharmacophore group looked at using a novel NIR probe, ZW760-mbc94 (51),[130] which contained a similar scaffold to NIR760, whereby the alkyl sulfonic acid groups were substituted with trimethyl amine cations. When compared with other ZW800-1, it contains a carbon–carbon linkage at the meso position as opposed to an enol ether.[153] The dye itself exhibited a fluorescence emission of 805 nm in DMSO, 786 nm in methanol, and 775 nm in H2O. When in the presence of a control ligand/blocking agent, 4-quinolone-3-carboxamide (4Q3C), the uptake of ZW760-mbc94 was lowered in CB2-mid DBT cells, with roughly ~50 % blockage. In WT DBT cells (non-CB2R expressing cells) the uptake of ZW760-mbc94 is ~40 % lower than of the CB2-mid DBT cells, demonstrating its specificity to CB2R. An in vitro fluorescence saturation binding study against 4Q3C gave a Kd = 53.9 nM when binding to CB2R. When compared with the previous NIR760-mbc94, the new ZW760 showed an improved binding specificity in vitro and ex vivo.
The same group developed IR700DX-mbc94 (52), utilising the NIR probe, IR700DX, as a potential phototherapeutic option for tumour cells overexpressing CB2R.[131] The compound displayed an absorbance and emission of 682 and 690 nm in methanol, respectively. IR700DX-mbc94 was determined to be non-toxic with the lack of irradiation, in both CHO-K1/CB2 and CB2-mid DBT cells. The compound displayed a Kd of 42.0 ± 19.6 nM and exhibited an ability to kill and inhibit CB2+ cells when irradiated, although requiring binding to CB2R. An in vivo targeted phototherapy study using IR700DX-mbc94 highlighted that 1O2 and free radicals were produced upon irradiation, producing a necrosis-like cell death.[154] IR700DX-mbc94 shows potential to be a selective phototherapy agent for cancer cells; it bares little to no cytotoxicity before irradiation and displays selectivity towards CB2+ cells. IR700DX-mbc94 was further modified to contain a dimethyl thioketal within the linker. The new probe, IR700DX-TK-mbc94 (53), was found to displace [3H]-CP 55,940 from hCB2R with nanomolar affinity, as did IR700DX-mbc94.[132] In CB2R+ mouse delayed brain tumour CB2-mid DBT cells, IR700DX-TK-mbc94 showed 59 % cell death upon irradiation whereas IR700DX-mbc94 showed negligible amounts. The higher percentage of cell death when treated with IR700DX-TK-mbc94 was consistent across varying the time and concentration of the incubation. HEK-293 cells exhibited much less cell death than CB2-mid DBT cells, indicating the potential of selective phototherapy agents for tumours expressing CB2R.
T1117 (54) was first reported in 2010 and derived from the CB1R inverse agonist, AM251, tagged with fluorescent moiety, 5-carboxytetramethylrhodamine (5-TAMRA).[133] AM251 displaced CP 55,940 from CB1R WT mouse brain membranes with a Ki of 0.8 nM, whereas T1117 only displayed 10 % displacement at 1 μM. T1117 also displayed binding to GPR55 through Ca+ response in HEK-293 cells overexpressing recombinant GPR55 and showed fluorescence in WT mouse mesenteric artery (MMA). A conflicting study in 2014 demonstrated T1117 binds to CB1R showcasing a Kd of 460 ± 80 nM.[134] In their fluorescence quenching study, T1117 displayed a decrease in fluorescence when incubated in mouse membranes when compared with the PBS control and an increase when AM251 was added. It was validated that fluorescence of T1117 was quenched upon binding to CB1R in overexpressing HEK-293 cells. T1117 binding to CB1R was shown to be sensitive to the NAM Org27569 and this fluorescence-quenching method has been proposed to be suitable for rapid assessment of novel orthosteric and allosteric modulators. Despite this, the ligand was opted to be not suitable for in situ CB1R identification due to non-specific membrane permeability. A study in 2019 focusing on linker attachments of SR141716A suggested that C-5 of the pharmacophore was a suitable position for fluorescent moiety linkage.[155] A fluorescein fluorophore showed affinity to hCB1R at 260 ± 20 nM with a linker length of 12 atoms, suggesting the C-5 position can be utilised for high affinity CB1R probes.
Fluorescent Oxoquinoline Derivatives
As one of the first fluorophores to be used for CB2R in vivo imaging, NIR760 dye was further coupled to another selective CB2R ligand. The quinoline scaffold had been shown to be a highly selective CB2R ligand with selectivity up to 0.2 nM.[156] The 4-quinoline-3-carboxamide with a pentyl at N-1 was the chosen scaffold for NIR760 coupling. NIR760-Q (55) was synthesised through reported steps to form 4Q3C and linked via a click reaction to form a 1,2,3-triazole with an ether bridge.[135] The novel probe, NIR760-Q, has an absorbance and emission of 768 and 787 nm, respectively, and a quantum yield of 16.5 % in water. Having similar absorbance and emission as NIRmbc94, the compound’s binding affinity was tested in Jurkat cells. These cells overexpress CB2R, whereas testing previously was done on CB2-mid DBT cells. NIR760 showed a Kd of 75.51 nM within an excess of 4Q3C to saturate binding sites. Cells with both 4Q3C and NIR760-Q had lower fluorescence than that of just the probe, indicating specific CB2R binding. The Jurkat cells incubated with NIR760-Q showed a ×2.8 increase over those just treated with NIR760. However, partial inhibition of the compound indicated non-specific binding, which was accounted for by the overall net charge of the compound.
A study proposed a new scaffold, pyrazolo[1,5-a]pyrimidine, for CB2R selective compounds boasting a similar structure to 4Q3C. With the large hydrophobic adamantane amide and the N-1 alkyl pentane, the para-substituted phenyl derivatives had high affinities to CB2R.[157] The substitution at the C-2 position has very little effect on the binding affinity with these compounds. Based on this, XLP4 was initially synthesised, containing a bromide handle at C-2 for further chemistry.[136] This compound underwent Suzuki cross coupling with 4-carboxyphenylboronic acid, followed by a 1,6-diaminohexane addition to form XLP6, which was coupled to NIR760 dye to yield the CB2R selective probe, NIR760-XLP6 (56). The probe has an absorbance and emission of 766 and 785 nm, respectively, and a quantum yield of 11.6 % in water, boasting similar photophysical properties to NIR760-Q. Saturated binding with XLP4 in DBT-CB2 cells, showed NIR760-XLP6 binding to CB2 with a Kd of 169.1 nM. Higher fluorescence was observed in the DBT-CB2 cells as opposed to the DBT-CB1 cells, confirming CB2R binding. The compound was found to have selectivity towards CB2R both in vitro and in vivo. Further tests on the CB2 overexpressing pancreatic duct adenocarcinoma cells (PDAC) found NIR760-XLP6 binds to CB2R in PNAC-1 with high imaging contrast in vivo after 9 h post injection and ex vivo up to 48 h with certain lymph nodes.[158]
The mbc94 and NIR760 probes (Q and XLP6) had proven to show good fluorescence properties in biological systems while maintaining moderate affinity to CB2R. Similar to NIR760-Q, the N-adamantyl-4-oxo-1,4-dihydroquinoline-3-carboxamide scaffold was selected due to CB2R affinity and its tolerance to bulky, aromatic groups at N-1.[159] Large fluorophores, 4-dimethylaminophthalimide (4-DMAP), NBD, and fluorescein-thiourea (FTU), were selected as the fluorescent moieties with the selective quinoline.[137] The linker for this study was a varying methylene chain from six to twelve carbons, in which a 6 chain-DMAP probe (57) was found have the highest affinity (Ki(hCB1R) > 10000 nM; Ki(hCB2R) = 130 nM). The emission of the DMAP fluorophore varied between 504 and 544 nm in various solvents, combined with ideal pharmacological properties, the probe was chosen for fluorescent-activated cell sorting (FACS). The results obtained from a fluoro-binding assay against WIN 55,212-2 and GW405833 matched literature values.[160] The compound displayed low cytotoxicity in numerous different cell lines within 24 h of incubation. A saturation binding assay in CB2R overexpressing HEK-293 cells displaced GW405833 with a Kd of 44 nM, while an absence of fluorescence was observed in WT HEK-293 cells. Several CB2R expressing cancer cell lines were tested with the fluorescent ligand and GW405833 in saturation binding assays, in which the results were consistent with the HEK-293 CB2R cell model. Fluorescence microscopy demonstrated the ideal concentration for imaging of the fluorescent ligand was 15 uM, with higher concentrations exhibiting off-site binding. The IC50 of GW405833 (IC50 = 3.94 nM) was obtained using the model and verified by comparison to the radioligand binding (Ki = 3.92 nM), validating the compounds ability to be applied as an option to conventional radioligand assays with CB2R specificity.
Similarly to the quinoline analogues, 1,8-naphthyridine compounds have shown to be highly selective CB2R compounds, while maintaining lower lipophilicity. Variations on the N-1, C-3, and C-6 positions have highlighted a tolerance for bulky groups at N-1, whereas substituents at C-6 affect functional activity, potentially reducing CB2R affinity. The receptor binding affinity of the carboxamide at C-3 can be altered using 4-methylcyclohexyl, with cis derivatives giving higher affinity than trans. In 2018, Cooper et al. modified the scaffold at N-1 with PEG and methylene linkers bound to a BODIPY630/650-X fluorophore.[138] These two compounds were screened by a radioligand competition binding assay displacing [3H]-CP 55,940. The previously reported N-ethyl morpholino analogue, used as a comparison, had an affinity similar to that of SR144528 (pKi(CB1R) < 5; pKi(CB2R) = 7.26 ± 0.04). The attachment of both linkers and BODIPY630/650-X resulted in a loss in CB2R affinity as well as inverse agonism due to substitution at C-6 (pKi(CB1R) < 5; pKi(CB2R) < 5). Due to a lack of affinity with the C-6 substitution, modifying the cyclohexyl group at C-3 with the fluorophore was performed. The only fluorescent ligand to have moderate CB2R affinity was BODIPY, in the cis conformer 58. This compound showed a similar pIC50 to that of SR144528 despite having a weaker affinity (IC50(CB2R) = 6.72 ± 0.18). Unfortunately, this compound lacked any beneficial fluorescent properties from cellular imaging and cells pre-incubated with SR14458 did not affect the labelling, displaying non-CB2R fluorescence.
Fluorescent Pyrazine/Pyridine Cannabinoids
The Carreira group focussed on modifying pyrazine and pyridine scaffolds, in which no previous cannabinoid fluorescent probes have been published.[139] With parent compounds RO6839251 (Ki(hCB2R) = 0.2 nM) and RO6852763 (Ki(hCB2R) = 0.2 nM), displaying good affinity and selectivity to CB2R, fluorescent moieties NBD, AttoThio12, Silicon Rhodamine (SiR), and Cy5.5 were linked via variations on PEG or C-6 alkyl chains. NBD (59) and AttoThio12 (60) linked compounds displayed the highest affinity to CB2R but maintained moderate affinity to CB1R. In flow cytometry experiments, AttoThio12 and SiR (61) bound compounds labelled mouse and human CB2R, CHO cells overexpressing cannabinoid receptors, as well as WT CHO cells. Specificity was further validated by displacing agonist, JWH133 and an inverse agonist, RO6851228. Utilising time-resolved FRET (TR-FRET), the values obtained in competitive binding against HU-308 and SR144528 with 61 supported the use of this methodology for CB2R high-throughput screening. Lastly, SiR was able to label hCB2R-overexpressing CHO cells, displaying no membrane labelling indicating CB2R specificity. This ultimately signifies a range of fluorescent probes that potentially can assist in elucidation of the CB2R mechanism of action.
Bifunctional Fluorescent Probes
A range of bifunctional CB compounds based of a hybrid between HU-308 and AM841 were synthesised by the Carreira group, containing a fluorescent or photoaffinity moiety and an electrophilic group.[140] The compounds were tested within the membrane of CHO cells overexpressing human cannabinoid receptors via a radioligand assay against [3H]-CP 55,940. Selectivity at hCB2R of these compounds ranged from 1 to > 2381, with NBD compounds having the highest potency. Amongst the NBD compounds, both the isothiocyano (62) and azido (63) had the highest hCB2R selectivity. It was noted that 63 did not covalently bind to hCB2R overexpressing CHO cells as per a polyacrylamide gel electrophoresis assay. Further establishing upon the scaffold, the fluorophore was altered to DY-480XL, Alexa647, and AttoThio12, while the terminal azido remained consistent.[141,142] From these compounds, the DY-480XL (64) and AttoThio12 (65) dyes demonstrated high affinity and selectivity to CB2R. Both probes were suggested to be alternatives to radioligands as per a TR-FRET assay. Notably, 65 could be detected in 5xFAD mice, expressing mCB2R AD microglia, and labelled endogenous hCB2R MDA-MB-231 breast cancer cells by flow cytometry. Finally, real-time confocal fluorescence microscopy displayed that 65 labelled mCB2R splenocytes and hCB2R macrophages with minimal internalisation, suggesting the probe can be used for endogenous CB2R in murine and human models.
Conclusions and Future Perspectives
Modulation of the endocannabinoid system is an ideal therapeutic target for a plethora of neurological diseases and anti-inflammatory effects, primarily focusing on CB2R selectivity. Modern drug discovery ultimately necessitates the capability to image the receptor and the demonstration of target engagement, to understand the mechanism of action in a complicated system, such as the ECS. With a multitude of screening assays and several potential off-target interactions, a more holistic and comprehensive screening approach can give a better view of the true activity spectrum.[33] Small molecular probes are a necessity for further drug development, in which the current trend in cannabinoid small molecule probes is shifted towards CB2R selective compounds with a fluorescent moiety. Within recent years, bifunctional probes applying the ability to covalently bind to the receptor, via an electrophile or photoactivatable group, have become more prominent. To determine and further elucidate CB2R pharmacology, selective scaffolds can be highly utilised for attachment of imaging moieties, specifically noting quinolone, indole, and thiophene derivatives, containing a large hydrophobic carboxamide.[161–163] For a consistent analysis of CB imaging compounds, altering the probe and/or position exclusively within the same scaffold could clarify binding affinity and efficacy, however this ultimately could prove to be a large task.
Ultimately, new small molecular probes can be designed for in vivo imaging, with the possibility of elucidating biological pathways and highlighting diseases for treatment and diagnosis within these pathways. Several issues have the potential to arise with in vivo testing, namely lipophilicity and specificity of the ligand once the fluorophore is linked. While most PET probes have minimal issues with blood–brain barrier permeation, the addition of a fluorophore tag could have negative effects on the physiochemical properties and possibly render neurological imaging difficult. Another issue is competitive clearance pathways, in which net-neutral charged fluorophores are suggested to avoid this rapid clearance issue.[164] Finally, the probe needs to have a high affinity to the receptor, to overcome non-selective binding, which is affected upon fluorophore coupling. Non-invasive probes utilising near-infrared and BODIPY dyes have the potential for in vivo imaging due to a negation of bioluminescence, although membrane permeability could still prove to be problematic.[165,166] With recent successes in cannabinoid imaging, the probes mentioned in this review can potentially further guide drug discovery and help uncover the sophisticated cannabinoid pharmacology.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
HG is grateful for the ARC DECRA DE160100482.
References
[1] S. P. Alexander, Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 157.| Crossref | GoogleScholarGoogle Scholar | 26216862PubMed |
[2] A. C. Campos, M. V. Fogaca, A. B. Sonego, F. S. Guimaraes, Pharmacol. Res. 2016, 112, 119.
| Crossref | GoogleScholarGoogle Scholar | 26845349PubMed |
[3] V. Di Marzo, M. Bifulco, L. De Petrocellis, Nat. Rev. Drug Discov. 2004, 3, 771.
| Crossref | GoogleScholarGoogle Scholar | 15340387PubMed |
[4] R. Mechoulam, Y. Shvo, I. Hashish, Tetrahedron 1963, 19, 2073.
| Crossref | GoogleScholarGoogle Scholar | 5879214PubMed |
[5] R. Mechoulam, Y. Gaoni, J. Am. Chem. Soc. 1965, 87, 3273.
| Crossref | GoogleScholarGoogle Scholar | 14324315PubMed |
[6] W. A. Devane, F. A. Dysarz, M. R. Johnson, L. S. Melvin, A. C. Howlett, Mol. Pharmacol. 1988, 34, 605.
| 2848184PubMed |
[7] M. Herkenham, A. B. Lynn, M. D. Little, M. R. Johnson, L. S. Melvin, B. R. de Costa, et al. Proc. Natl. Acad. Sci. USA 1990, 87, 1932.
| Crossref | GoogleScholarGoogle Scholar | 2308954PubMed |
[8] L. A. Matsuda, S. J. Lolait, M. J. Brownstein, A. C. Young, T. I. Bonner, Nature 1990, 346, 561.
| Crossref | GoogleScholarGoogle Scholar | 2165569PubMed |
[9] S. Munro, K. L. Thomas, M. Abu-Shaar, Nature 1993, 365, 61.
| Crossref | GoogleScholarGoogle Scholar | 7689702PubMed |
[10] W. A. Devane, L. Hanus, A. Breuer, R. G. Pertwee, L. A. Stevenson, G. Griffin, et al. Science 1992, 258, 1946.
| Crossref | GoogleScholarGoogle Scholar | 1470919PubMed |
[11] T. Sugiura, S. Kondo, A. Sukagawa, S. Nakane, A. Shinoda, K. Itoh, et al. Biochem. Biophys. Res. Commun. 1995, 215, 89.
| Crossref | GoogleScholarGoogle Scholar | 7575630PubMed |
[12] R. Mechoulam, S. Ben-Shabat, L. Hanus, M. Ligumsky, N. E. Kaminski, A. R. Schatz, et al. Biochem. Pharmacol. 1995, 50, 83.
| Crossref | GoogleScholarGoogle Scholar | 7605349PubMed |
[13] R. G. Pertwee, in Cannabinoids: Handbook of Experimental Pharmacology (Ed. R. G. Pertwee) 2005, Vol. 168, pp. 1–51 (Springer: Berlin).
[14] Y. Okamoto, J. Morishita, K. Tsuboi, T. Tonai, N. Ueda, J. Biol. Chem. 2004, 279, 5298.
| Crossref | GoogleScholarGoogle Scholar | 14634025PubMed |
[15] B. F. Cravatt, D. K. Giang, S. P. Mayfield, D. L. Boger, R. A. Lerner, N. B. Gilula, Nature 1996, 384, 83.
| Crossref | GoogleScholarGoogle Scholar | 8900284PubMed |
[16] V. Di Marzo, L. De Petrocellis, T. Bisogno, Handb. Exp. Pharmacol. 2005, 168, 147.
| Crossref | GoogleScholarGoogle Scholar |
[17] M. Reisenberg, P. K. Singh, G. Williams, P. Doherty, Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3264.
| Crossref | GoogleScholarGoogle Scholar | 23108545PubMed |
[18] J. L. Blankman, G. M. Simon, B. F. Cravatt, Chem. Biol. 2007, 14, 1347.
| Crossref | GoogleScholarGoogle Scholar | 18096503PubMed |
[19] D. K. Nomura, B. E. Morrison, J. L. Blankman, J. Z. Long, S. G. Kinsey, M. C. Marcondes, et al. Science 2011, 334, 809.
| Crossref | GoogleScholarGoogle Scholar | 22021672PubMed |
[20] A. H. Lichtman, D. Leung, C. C. Shelton, A. Saghatelian, C. Hardouin, D. L. Boger, et al. J. Pharmacol. Exp. Ther. 2004, 311, 441.
| Crossref | GoogleScholarGoogle Scholar | 15229230PubMed |
[21] S. G. Kinsey, J. Z. Long, S. T. O’Neal, R. A. Abdullah, J. L. Poklis, D. L. Boger, et al. J. Pharmacol. Exp. Ther. 2009, 330, 902.
| Crossref | GoogleScholarGoogle Scholar | 19502530PubMed |
[22] L. Cristino, T. Bisogno, V. Di Marzo, Nat. Rev. Neurol. 2020, 16, 9.
| Crossref | GoogleScholarGoogle Scholar | 31831863PubMed |
[23] Z. Shao, J. Yin, K. Chapman, M. Grzemska, L. Clark, J. Wang, et al. Nature 2016, 540, 602.
| Crossref | GoogleScholarGoogle Scholar | 27851727PubMed |
[24] T. Hua, K. Vemuri, M. Pu, L. Qu, G. W. Han, Y. Wu, et al. Cell 2016, 167, 750.
| Crossref | GoogleScholarGoogle Scholar | 27768894PubMed |
[25] X. Li, T. Hua, K. Vemuri, J. H. Ho, Y. Wu, L. Wu, et al. Cell 2019, 176, 459.
| Crossref | GoogleScholarGoogle Scholar | 30639103PubMed |
[26] E. Ryberg, N. Larsson, S. Sjogren, S. Hjorth, N. O. Hermansson, J. Leonova, et al. Br. J. Pharmacol. 2007, 152, 1092.
| Crossref | GoogleScholarGoogle Scholar | 17876302PubMed |
[27] A. J. Brown, Br. J. Pharmacol. 2007, 152, 567.
| Crossref | GoogleScholarGoogle Scholar | 17906678PubMed |
[28] M. Kohno, H. Hasegawa, A. Inoue, M. Muraoka, T. Miyazaki, K. Oka, et al. Biochem. Biophys. Res. Commun. 2006, 347, 827.
| Crossref | GoogleScholarGoogle Scholar | 16844083PubMed |
[29] D. McHugh, Br. J. Pharmacol. 2012, 167, 1575.
| Crossref | GoogleScholarGoogle Scholar | 22563843PubMed |
[30] D. McHugh, J. Page, E. Dunn, H. B. Bradshaw, Br. J. Pharmacol. 2012, 165, 2414.
| Crossref | GoogleScholarGoogle Scholar | 21595653PubMed |
[31] V. Di Marzo, Nat. Rev. Drug Discov. 2018, 17, 623.
| Crossref | GoogleScholarGoogle Scholar | 30116049PubMed |
[32] F. A. Iannotti, C. L. Hill, A. Leo, A. Alhusaini, C. Soubrane, E. Mazzarella, et al. ACS Chem. Neurosci. 2014, 5, 1131.
| Crossref | GoogleScholarGoogle Scholar | 25029033PubMed |
[33] M. Soethoudt, U. Grether, J. Fingerle, T. W. Grim, F. Fezza, L. de Petrocellis, et al. Nat. Commun. 2017, 8, 13958.
| Crossref | GoogleScholarGoogle Scholar | 28045021PubMed |
[34] S. E. Turner, C. M. Williams, L. Iversen, B. J. Whalley, Prog. Chem. Org. Nat. Prod. 2017, 103, 61.
| Crossref | GoogleScholarGoogle Scholar | 28120231PubMed |
[35] E. S. Onaivi, Curr. Neuropharmacol. 2011, 9, 205.
| Crossref | GoogleScholarGoogle Scholar | 21886591PubMed |
[36] L. Iversen, Brain 2003, 126, 1252.
| Crossref | GoogleScholarGoogle Scholar | 12764049PubMed |
[37] S. N. Davies, R. G. Pertwee, G. Riedel, Neuropharmacology 2002, 42, 993.
| Crossref | GoogleScholarGoogle Scholar | 12128000PubMed |
[38] J. E. Kuster, J. I. Stevenson, S. J. Ward, T. E. D’Ambra, D. A. Haycock, J. Pharmacol. Exp. Ther. 1993, 264, 1352.
| 8450470PubMed |
[39] N. Uchiyama, M. Kawamura, R. Kikura-Hanajiri, Y. Goda, Forensic Toxicol. 2012, 30, 114.
| Crossref | GoogleScholarGoogle Scholar |
[40] S. D. Banister, S. M. Wilkinson, M. Longworth, J. Stuart, N. Apetz, K. English, et al. ACS Chem. Neurosci. 2013, 4, 1081.
| Crossref | GoogleScholarGoogle Scholar | 23551277PubMed |
[41] A. Blasio, A. Iemolo, V. Sabino, S. Petrosino, L. Steardo, K. C. Rice, et al. Neuropsychopharmacology 2013, 38, 2498.
| Crossref | GoogleScholarGoogle Scholar | 23793355PubMed |
[42] R. Christensen, P. K. Kristensen, E. M. Bartels, H. Bliddal, A. Astrup, Lancet 2007, 370, 1706.
| Crossref | GoogleScholarGoogle Scholar | 18022033PubMed |
[43] M. Rinaldi-Carmona, F. Barth, M. Héaulme, D. Shire, B. Calandra, C. Congy, et al. FEBS Lett. 1994, 350, 240.
| Crossref | GoogleScholarGoogle Scholar | 8070571PubMed |
[44] A. H. Sam, V. Salem, M. A. Ghatei, J. Obes. 2011, 2011, 432607.
| Crossref | GoogleScholarGoogle Scholar | 21773005PubMed |
[45] A. A. Izzo, F. Borrelli, R. Capasso, V. Di Marzo, R. Mechoulam, Trends Pharmacol. Sci. 2009, 30, 515.
| Crossref | GoogleScholarGoogle Scholar | 19729208PubMed |
[46] A. J. Hill, C. M. Williams, B. J. Whalley, G. J. Stephens, Pharmacol. Ther. 2012, 133, 79.
| Crossref | GoogleScholarGoogle Scholar | 21924288PubMed |
[47] T. A. Reekie, M. P. Scott, M. Kassiou, Nat. Rev. Chem. 2018, 2, 0101.
| Crossref | GoogleScholarGoogle Scholar |
[48] O. Devinsky, M. R. Cilio, H. Cross, J. Fernandez-Ruiz, J. French, C. Hill, et al. Epilepsia 2014, 55, 791.
| Crossref | GoogleScholarGoogle Scholar | 24854329PubMed |
[49] R. Mechoulam, M. Peters, E. Murillo-Rodriguez, L. O. Hanus, Chem. Biodivers. 2007, 4, 1678.
| Crossref | GoogleScholarGoogle Scholar | 17712814PubMed |
[50] F. F. Peres, A. C. Lima, J. E. C. Hallak, J. A. Crippa, R. H. Silva, V. C. Abilio, Front. Pharmacol. 2018, 9, 482.
| Crossref | GoogleScholarGoogle Scholar | 29867488PubMed |
[51] R. B. Laprairie, A. M. Bagher, M. E. Kelly, E. M. Denovan-Wright, Br. J. Pharmacol. 2015, 172, 4790.
| Crossref | GoogleScholarGoogle Scholar | 26218440PubMed |
[52] M. Tham, O. Yilmaz, M. Alaverdashvili, M. E. M. Kelly, E. M. Denovan-Wright, R. B. Laprairie, Br. J. Pharmacol. 2019, 176, 1455.
| Crossref | GoogleScholarGoogle Scholar | 29981240PubMed |
[53] L. Khurana, K. Mackie, D. Piomelli, D. A. Kendall, Neuropharmacology 2017, 124, 3.
| Crossref | GoogleScholarGoogle Scholar | 28527758PubMed |
[54] G. Navarro, P. Morales, C. Rodriguez-Cueto, J. Fernandez-Ruiz, N. Jagerovic, R. Franco, Front. Neurosci. 2016, 10, 406.
| Crossref | GoogleScholarGoogle Scholar | 27679556PubMed |
[55] S. Hryhorowicz, M. Kaczmarek-Rys, A. Andrzejewska, K. Staszak, M. Hryhorowicz, A. Korcz, et al. Int. J. Mol. Sci. 2019, 20, 5874.
| Crossref | GoogleScholarGoogle Scholar |
[56] B. G. Ramirez, C. Blazquez, T. Gomez del Pulgar, M. Guzman, M. L. de Ceballos, J. Neurosci. 2005, 25, 1904.
| Crossref | GoogleScholarGoogle Scholar | 15728830PubMed |
[57] J. Ehrhart, D. Obregon, T. Mori, H. Hou, N. Sun, Y. Bai, et al. J. Neuroinflammation 2005, 2, 29.
| Crossref | GoogleScholarGoogle Scholar | 16343349PubMed |
[58] A. M. Martin-Moreno, D. Reigada, B. G. Ramirez, R. Mechoulam, N. Innamorato, A. Cuadrado, et al. Mol. Pharmacol. 2011, 79, 964.
| Crossref | GoogleScholarGoogle Scholar | 21350020PubMed |
[59] J. G. Zarruk, D. Fernandez-Lopez, I. Garcia-Yebenes, M. S. Garcia-Gutierrez, J. Vivancos, F. Nombela, et al. Stroke 2012, 43, 211.
| Crossref | GoogleScholarGoogle Scholar | 22020035PubMed |
[60] R. G. Pertwee, Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3353.
| Crossref | GoogleScholarGoogle Scholar | 23108552PubMed |
[61] S. Han, J. Thatte, D. J. Buzard, R. M. Jones, J. Med. Chem. 2013, 56, 8224.
| Crossref | GoogleScholarGoogle Scholar | 23865723PubMed |
[62] G. M. Simon, M. J. Niphakis, B. F. Cravatt, Nat. Chem. Biol. 2013, 9, 200.
| Crossref | GoogleScholarGoogle Scholar | 23508173PubMed |
[63] J. A. Wagner, Annu. Rev. Pharmacol. Toxicol. 2008, 48, 631.
| Crossref | GoogleScholarGoogle Scholar | 17937595PubMed |
[64] L. Zuurman, A. E. Ippel, E. Moin, J. M. van Gerven, Br. J. Clin. Pharmacol. 2009, 67, 5.
| Crossref | GoogleScholarGoogle Scholar | 19133057PubMed |
[65] A. Minichino, M. Senior, N. Brondino, S. H. Zhang, B. R. Godwlewska, P. W. J. Burnet, et al. J. Am. Med. Assoc. Psychiatry 2019, 76, 914.
| Crossref | GoogleScholarGoogle Scholar |
[66] F. Navarrete, M. S. Garcia-Gutierrez, R. Jurado-Barba, G. Rubio, A. Gasparyan, A. Austrich-Olivares, et al. Front. Psychiatry 2020, 11, 315.
| Crossref | GoogleScholarGoogle Scholar | 32395111PubMed |
[67] M. E. Bunnage, E. L. Chekler, L. H. Jones, Nat. Chem. Biol. 2013, 9, 195.
| Crossref | GoogleScholarGoogle Scholar | 23508172PubMed |
[68] P. M. Matthews, E. A. Rabiner, J. Passchier, R. N. Gunn, Br. J. Clin. Pharmacol. 2012, 73, 175.
| Crossref | GoogleScholarGoogle Scholar | 21838787PubMed |
[69] T. Binderup, U. Knigge, A. Loft, B. Federspiel, A. Kjaer, Clin. Cancer Res. 2010, 16, 978.
| Crossref | GoogleScholarGoogle Scholar | 20103666PubMed |
[70] B. D. Cheson, R. I. Fisher, S. F. Barrington, F. Cavalli, L. H. Schwartz, E. Zucca, et al. J. Clin. Oncol. 2014, 32, 3059.
| Crossref | GoogleScholarGoogle Scholar | 25113753PubMed |
[71] N. Ghosh, O. E. Rimoldi, R. S. Beanlands, P. G. Camici, Eur. Heart J. 2010, 31, 2984.
| Crossref | GoogleScholarGoogle Scholar | 20965888PubMed |
[72] C. R. Jack, D. S. Knopman, W. J. Jagust, L. M. Shaw, P. S. Aisen, M. W. Weiner, et al. Lancet Neurol. 2010, 9, 119.
| Crossref | GoogleScholarGoogle Scholar | 20083042PubMed |
[73] N. Evens, G. M. Bormans, Curr. Top. Med. Chem. 2010, 10, 1527.
| Crossref | GoogleScholarGoogle Scholar | 20583994PubMed |
[74] A. Cooper, S. Singh, S. Hook, J. D. A. Tyndall, A. J. Vernall, Pharmacol. Rev. 2017, 69, 316.
| Crossref | GoogleScholarGoogle Scholar | 28655732PubMed |
[75] M. Schurmann, P. Janning, S. Ziegler, H. Waldmann, Cell Chem. Biol. 2016, 23, 435.
| Crossref | GoogleScholarGoogle Scholar | 27049669PubMed |
[76] L. A. Stoddart, E. K. M. Johnstone, A. J. Wheal, J. Goulding, M. B. Robers, T. Machleidt, et al. Nat. Methods 2015, 12, 661.
| Crossref | GoogleScholarGoogle Scholar | 26030448PubMed |
[77] L. A. Stoddart, C. W. White, K. Nguyen, S. J. Hill, K. D. Pfleger, Br. J. Pharmacol. 2016, 173, 3028.
| Crossref | GoogleScholarGoogle Scholar | 26317175PubMed |
[78] P. P. Geurink, L. M. Prely, G. A. van der Marel, R. Bischoff, H. S. Overkleeft, Top. Curr. Chem. 2011, 324, 85.
| Crossref | GoogleScholarGoogle Scholar |
[79] E. Smith, I. Collins, Future Med. Chem. 2015, 7, 159.
| Crossref | GoogleScholarGoogle Scholar | 25686004PubMed |
[80] J. Sumranjit, S. J. Chung, Molecules 2013, 18, 10425.
| Crossref | GoogleScholarGoogle Scholar | 23994969PubMed |
[81] D. J. Lapinsky, D. S. Johnson, Future Med. Chem. 2015, 7, 2143.
| Crossref | GoogleScholarGoogle Scholar | 26511756PubMed |
[82] A. Charalambous, G. Yan, D. B. Houston, A. C. Howlett, D. R. Compton, B. R. Martin, et al. J. Med. Chem. 1992, 35, 3076.
| Crossref | GoogleScholarGoogle Scholar | 1323683PubMed |
[83] S. H. Burstein, C. A. Audette, A. Charalambous, S. A. Doyle, Y. Guo, S. A. Hunter, et al. Biochem. Biophys. Res. Commun. 1991, 176, 492.
| Crossref | GoogleScholarGoogle Scholar | 1850270PubMed |
[84] Y. Guo, V. Abadji, K. L. Morse, D. J. Fournier, X. Li, A. Makriyannis, J. Med. Chem. 1994, 37, 3867.
| Crossref | GoogleScholarGoogle Scholar | 7966145PubMed |
[85] S. L. Palmer, G. A. Thakur, A. Makriyannis, Chem. Phys. Lipids 2002, 121, 3.
| Crossref | GoogleScholarGoogle Scholar | 12505686PubMed |
[86] K. L. Morse, D. J. Fournier, X. Li, J. Grzybowska, A. Makriyannis, Life Sci. 1995, 56, 1957.
| Crossref | GoogleScholarGoogle Scholar | 7776819PubMed |
[87] A. D. Khanolkar, S. L. Palmer, A. Makriyannis, Chem. Phys. Lipids 2000, 108, 37.
| Crossref | GoogleScholarGoogle Scholar | 11106781PubMed |
[88] C. Chu, A. Ramamurthy, A. Makriyannis, M. A. Tius, J. Org. Chem. 2003, 68, 55.
| Crossref | GoogleScholarGoogle Scholar | 12515461PubMed |
[89] R. P. Picone, A. D. Khanolkar, W. Xu, L. A. Ayotte, G. A. Thakur, D. P. Hurst, et al. Mol. Pharmacol. 2005, 68, 1623.
| Crossref | GoogleScholarGoogle Scholar | 16157695PubMed |
[90] Y. Pei, R. W. Mercier, J. K. Anday, G. A. Thakur, A. M. Zvonok, D. Hurst, et al. Chem. Biol. 2008, 15, 1207.
| Crossref | GoogleScholarGoogle Scholar | 19022181PubMed |
[91] H. Zhou, Y. Peng, A. Halikhedkar, P. Fan, D. R. Janero, G. A. Thakur, et al. ACS Chem. Neurosci. 2017, 8, 1338.
| Crossref | GoogleScholarGoogle Scholar | 28220706PubMed |
[92] D. D. Dixon, M. A. Tius, G. A. Thakur, H. Zhou, A. L. Bowman, V. G. Shukla, et al. Bioorg. Med. Chem. Lett. 2012, 22, 5322.
| Crossref | GoogleScholarGoogle Scholar | 22796181PubMed |
[93] G. Ogawa, M. A. Tius, H. Zhou, S. P. Nikas, A. Halikhedkar, S. Mallipeddi, et al. J. Med. Chem. 2015, 58, 3104.
| Crossref | GoogleScholarGoogle Scholar | 25760146PubMed |
[94] R. W. Mercier, Y. Pei, L. Pandarinathan, D. R. Janero, J. Zhang, A. Makriyannis, Chem. Biol. 2010, 17, 1132.
| Crossref | GoogleScholarGoogle Scholar | 21035736PubMed |
[95] K. Yamada, K. C. Rice, J. L. Flippen-Anderson, M. A. Eissenstat, S. J. Ward, M. R. Johnson, et al. J. Med. Chem. 1996, 39, 1967.
| Crossref | GoogleScholarGoogle Scholar | 8642555PubMed |
[96] P. M. Kulkarni, A. R. Kulkarni, A. Korde, R. B. Tichkule, R. B. Laprairie, E. M. Denovan-Wright, et al. J. Med. Chem. 2016, 59, 44.
| Crossref | GoogleScholarGoogle Scholar | 26529344PubMed |
[97] R. B. Laprairie, A. R. Kulkarni, P. M. Kulkarni, D. P. Hurst, D. Lynch, P. H. Reggio, et al. ACS Chem. Neurosci. 2016, 7, 776.
| Crossref | GoogleScholarGoogle Scholar | 27046127PubMed |
[98] S. R. Fernando, R. G. Pertwee, Br. J. Pharmacol. 1997, 121, 1716.
| Crossref | GoogleScholarGoogle Scholar | 9283708PubMed |
[99] C. Li, W. Xu, S. K. Vadivel, P. Fan, A. Makriyannis, J. Med. Chem. 2005, 48, 6423.
| Crossref | GoogleScholarGoogle Scholar | 16190768PubMed |
[100] D. R. Janero, S. Yaddanapudi, N. Zvonok, K. V. Subramanian, V. G. Shukla, E. Stahl, et al. ACS Chem. Neurosci. 2015, 6, 1400.
| Crossref | GoogleScholarGoogle Scholar | 25978068PubMed |
[101] L. Balas, M. G. Cascio, V. Di Marzo, T. Durand, Bioorg. Med. Chem. Lett. 2006, 16, 3765.
| Crossref | GoogleScholarGoogle Scholar | 16682198PubMed |
[102] A. Makriyannis, J. Med. Chem. 2014, 57, 3891.
| Crossref | GoogleScholarGoogle Scholar | 24707904PubMed |
[103] M. Soethoudt, G. Alachouzos, E. J. van Rooden, M. D. Moya-Garzon, R. van den Berg, L. H. Heitman, et al. Cannabis Cannabinoid Res. 2018, 3, 136.
| Crossref | GoogleScholarGoogle Scholar | 29992186PubMed |
[104] M. Soethoudt, S. C. Stolze, M. V. Westphal, L. van Stralen, A. Martella, E. J. van Rooden, et al. J. Am. Chem. Soc. 2018, 140, 6067.
| Crossref | GoogleScholarGoogle Scholar | 29420021PubMed |
[105] D. R. Janero, A. Korde, A. Makriyannis, Methods Enzymol. 2017, 593, 217.
| Crossref | GoogleScholarGoogle Scholar | 28750804PubMed |
[106] D. W. Szymanski, M. Papanastasiou, K. Melchior, N. Zvonok, R. W. Mercier, D. R. Janero, et al. J. Proteome Res. 2011, 10, 4789.
| Crossref | GoogleScholarGoogle Scholar | 21861534PubMed |
[107] J. Fichna, M. Bawa, G. A. Thakur, R. Tichkule, A. Makriyannis, D. M. McCafferty, et al. PLoS One 2014, 9, e109115.
| Crossref | GoogleScholarGoogle Scholar | 25541946PubMed |
[108] C. M. Keenan, M. A. Storr, G. A. Thakur, J. T. Wood, J. Wager-Miller, A. Straiker, et al. Br. J. Pharmacol. 2015, 172, 2406.
| Crossref | GoogleScholarGoogle Scholar | 25572435PubMed |
[109] S. Mallipeddi, S. Kreimer, N. Zvonok, K. Vemuri, B. L. Karger, A. R. Ivanov, et al. J. Proteome Res. 2017, 16, 2419.
| Crossref | GoogleScholarGoogle Scholar | 28374590PubMed |
[110] P. Keov, P. M. Sexton, A. Christopoulos, Neuropharmacology 2011, 60, 24.
| Crossref | GoogleScholarGoogle Scholar | 20637785PubMed |
[111] L. T. May, K. Leach, P. M. Sexton, A. Christopoulos, Annu. Rev. Pharmacol. Toxicol. 2007, 47, 1.
| Crossref | GoogleScholarGoogle Scholar | 17009927PubMed |
[112] M. R. Price, G. L. Baillie, A. Thomas, L. A. Stevenson, M. Easson, R. Goodwin, et al. Mol. Pharmacol. 2005, 68, 1484.
| Crossref | GoogleScholarGoogle Scholar | 16113085PubMed |
[113] D. G. Deutsch, R. Omeir, G. Arreaza, D. Salehani, G. D. Prestwich, Z. Huang, et al. Biochem. Pharmacol. 1997, 53, 255.
| Crossref | GoogleScholarGoogle Scholar | 9065728PubMed |
[114] L. Balas, T. Durand, S. Saha, I. Johnson, S. Mukhopadhyay, J. Med. Chem. 2009, 52, 1005.
| Crossref | GoogleScholarGoogle Scholar | 19161308PubMed |
[115] S. J. Briddon, B. Kellam, S. J. Hill, Methods Mol. Biol. 2011, 746, 211.
| Crossref | GoogleScholarGoogle Scholar | 21607859PubMed |
[116] A. J. Vernall, S. J. Hill, B. Kellam, Br. J. Pharmacol. 2014, 171, 1073.
| Crossref | GoogleScholarGoogle Scholar | 23734587PubMed |
[117] L. A. Stoddart, L. E. Kilpatrick, S. J. Briddon, S. J. Hill, Neuropharmacology 2015, 98, 48.
| Crossref | GoogleScholarGoogle Scholar | 25979488PubMed |
[118] R. J. Middleton, B. Kellam, Curr. Opin. Chem. Biol. 2005, 9, 517.
| Crossref | GoogleScholarGoogle Scholar | 16125436PubMed |
[119] S. Luo, E. Zhang, Y. Su, T. Cheng, C. Shi, Biomaterials 2011, 32, 7127.
| Crossref | GoogleScholarGoogle Scholar | 21724249PubMed |
[120] R. Weissleder, Nat. Biotechnol. 2001, 19, 316.
| Crossref | GoogleScholarGoogle Scholar | 11283581PubMed |
[121] C. Iliopoulos-Tsoutsouvas, R. N. Kulkarni, A. Makriyannis, S. P. Nikas, Expert Opin. Drug Discov. 2018, 13, 933.
| Crossref | GoogleScholarGoogle Scholar | 30249143PubMed |
[122] L. Martin-Couce, M. Martin-Fontecha, S. Capolicchio, M. L. Lopez-Rodriguez, S. Ortega-Gutierrez, J. Med. Chem. 2011, 54, 5265.
| Crossref | GoogleScholarGoogle Scholar | 21675776PubMed |
[123] L. Martin-Couce, M. Martin-Fontecha, O. Palomares, L. Mestre, A. Cordomi, M. Hernangomez, et al. Angew. Chem. Int. Ed. Engl. 2012, 51, 6896.
| Crossref | GoogleScholarGoogle Scholar | 22689411PubMed |
[124] M. Martin-Fontecha, A. Angelina, B. Ruckert, A. Rueda-Zubiaurre, L. Martin-Cruz, W. van de Veen, et al. Bioconjug. Chem. 2018, 29, 382.
| Crossref | GoogleScholarGoogle Scholar | 29314831PubMed |
[125] S. Singh, C. R. M. Oyagawa, C. Macdonald, N. L. Grimsey, M. Glass, A. J. Vernall, ACS Med. Chem. Lett. 2019, 10, 209.
| Crossref | GoogleScholarGoogle Scholar | 30783505PubMed |
[126] A. S. Yates, S. W. Doughty, D. A. Kendall, B. Kellam, Bioorg. Med. Chem. Lett. 2005, 15, 3758.
| Crossref | GoogleScholarGoogle Scholar | 15993070PubMed |
[127] R. R. Petrov, M. E. Ferrini, Z. Jaffar, C. M. Thompson, K. Roberts, P. Diaz, Bioorg. Med. Chem. Lett. 2011, 21, 5859.
| Crossref | GoogleScholarGoogle Scholar | 21855337PubMed |
[128] M. Sexton, G. Woodruff, E. A. Horne, Y. H. Lin, G. G. Muccioli, M. Bai, et al. Chem. Biol. 2011, 18, 563.
| Crossref | GoogleScholarGoogle Scholar | 21609837PubMed |
[129] S. Zhang, P. Shao, M. Bai, Bioconjug. Chem. 2013, 24, 1907.
| Crossref | GoogleScholarGoogle Scholar | 24094147PubMed |
[130] Z. Wu, P. Shao, S. Zhang, M. Bai, J. Biomed. Opt. 2014, 19, 036006.
| Crossref | GoogleScholarGoogle Scholar | 25364950PubMed |
[131] S. Zhang, N. Jia, P. Shao, Q. Tong, X. Q. Xie, M. Bai, Chem. Biol. 2014, 21, 338.
| Crossref | GoogleScholarGoogle Scholar | 24583052PubMed |
[132] X. Ling, S. Zhang, Y. Liu, M. Bai, J. Biomed. Opt. 2018, 23, 108001.
| Crossref | GoogleScholarGoogle Scholar |
[133] C. J. Daly, R. A. Ross, J. Whyte, C. M. Henstridge, A. J. Irving, J. C. McGrath, Br. J. Pharmacol. 2010, 159, 787.
| Crossref | GoogleScholarGoogle Scholar | 20136833PubMed |
[134] A. Bruno, F. Lembo, E. Novellino, M. Stornaiuolo, L. Marinelli, Sci. Rep. 2014, 4, 3757.
| Crossref | GoogleScholarGoogle Scholar | 24441508PubMed |
[135] Z. Wu, P. Shao, S. Zhang, X. Ling, M. Bai, J. Biomed. Opt. 2014, 19, 076016.
| Crossref | GoogleScholarGoogle Scholar | 25364950PubMed |
[136] X. Ling, S. Zhang, P. Shao, W. Li, L. Yang, Y. Ding, et al. Biomaterials 2015, 57, 169.
| Crossref | GoogleScholarGoogle Scholar | 25916505PubMed |
[137] F. Spinelli, R. Giampietro, A. Stefanachi, C. Riganti, J. Kopecka, F. S. Abatematteo, et al. Eur. J. Med. Chem. 2020, 188, 112037.
| Crossref | GoogleScholarGoogle Scholar | 31954990PubMed |
[138] A. G. Cooper, C. R. M. Oyagawa, J. J. Manning, S. Singh, S. Hook, N. L. Grimsey, et al. MedChemComm 2018, 9, 2055.
| Crossref | GoogleScholarGoogle Scholar | 30647881PubMed |
[139] T Gazzi, B Brennecke, K Atz, C Korn, D Sykes, R. C. Sarott, et al. ChemRxiv 2019, Preprint
| Crossref | GoogleScholarGoogle Scholar |
[140] M. V. Westphal, R. C. Sarott, E. A. Zirwes, A. Osterwald, W. Guba, C. Ullmer, et al. Chem. – Eur. J. 2020, 26, 1380.
| Crossref | GoogleScholarGoogle Scholar | 31961047PubMed |
[141] R Sarott, M Westphal, P Pfaff, C Korn, D Sykes, T Gazzi, et al. J. Am. Chem. Soc. 2020, 142, 16953.
| Crossref | GoogleScholarGoogle Scholar | 32902974PubMed |
[142] R Sarott, M Westphal, P Pfaff, C Korn, D Sykes, T Gazzi, et al. ChemRxiv 2019, Preprint.
| Crossref | GoogleScholarGoogle Scholar |
[143] V. P. Hytonen, Cell Chem. Biol. 2017, 24, 921.
| Crossref | GoogleScholarGoogle Scholar | 28820961PubMed |
[144] F. Fezza, S. Oddi, M. Di Tommaso, C. De Simone, C. Rapino, N. Pasquariello, et al. J. Lipid Res. 2008, 49, 1216.
| Crossref | GoogleScholarGoogle Scholar | 18316795PubMed |
[145] N. Titishov, R. Mechoulam, A. M. Zimmerman, Pharmacology 1989, 39, 337.
| Crossref | GoogleScholarGoogle Scholar | 2561381PubMed |
[146] L. Hanus, A. Breuer, S. Tchilibon, S. Shiloah, D. Goldenberg, M. Horowitz, et al. Proc. Natl. Acad. Sci. USA 1999, 96, 14228.
| Crossref | GoogleScholarGoogle Scholar | 10588688PubMed |
[147] J. Cumella, L. Hernandez-Folgado, R. Giron, E. Sanchez, P. Morales, D. P. Hurst, et al. ChemMedChem 2012, 7, 452.
| Crossref | GoogleScholarGoogle Scholar | 22302767PubMed |
[148] P. Morales, M. Gomez-Canas, G. Navarro, D. P. Hurst, F. J. Carrillo-Salinas, L. Lagartera, et al. J. Med. Chem. 2016, 59, 6753.
| Crossref | GoogleScholarGoogle Scholar | 27309150PubMed |
[149] M. Onoda, S. Uchiyama, T. Santa, K. Imai, Luminescence 2002, 17, 11.
| Crossref | GoogleScholarGoogle Scholar | 11816057PubMed |
[150] A. G. Cooper, C. MacDonald, M. Glass, S. Hook, J. D. A. Tyndall, A. J. Vernall, Eur. J. Med. Chem. 2018, 145, 770.
| Crossref | GoogleScholarGoogle Scholar | 29407590PubMed |
[151] M. Rinaldi-Carmona, F. Barth, J. Millan, J.-M. Derocq, P. Casellas, C. Congy, et al. J. Pharmacol. Exp. Ther. 1998, 284, 644.
| 9454810PubMed |
[152] M. Bai, M. Sexton, N. Stella, D. J. Bornhop, Bioconjug. Chem. 2008, 19, 988.
| Crossref | GoogleScholarGoogle Scholar | 18444670PubMed |
[153] H. S. Choi, S. L. Gibbs, J. H. Lee, S. H. Kim, Y. Ashitate, F. Liu, et al. Nat. Biotechnol. 2013, 31, 148.
| Crossref | GoogleScholarGoogle Scholar | 23292608PubMed |
[154] N. Jia, S. Zhang, P. Shao, C. Bagia, J. M. Janjic, Y. Ding, et al. Mol. Pharm. 2014, 11, 1919.
| Crossref | GoogleScholarGoogle Scholar | 24779700PubMed |
[155] P. S. Grant, N. Kahlcke, K. Govindpani, M. Hunter, C. MacDonald, M. A. Brimble, et al. Bioorg. Med. Chem. Lett. 2019, 29, 126644.
| Crossref | GoogleScholarGoogle Scholar | 31564385PubMed |
[156] S. Pasquini, M. De Rosa, V. Pedani, C. Mugnaini, F. Guida, L. Luongo, et al. J. Med. Chem. 2011, 54, 5444.
| Crossref | GoogleScholarGoogle Scholar | 21702498PubMed |
[157] M. Aghazadeh Tabrizi, P. G. Baraldi, G. Saponaro, A. R. Moorman, R. Romagnoli, D. Preti, et al. J. Med. Chem. 2013, 56, 4482.
| Crossref | GoogleScholarGoogle Scholar | 23697626PubMed |
[158] X. Guo, X. Ling, F. Du, Q. Wang, W. Huang, Z. Wang, et al. Transl. Oncol. 2018, 11, 1065.
| Crossref | GoogleScholarGoogle Scholar | 30005208PubMed |
[159] S. Pasquini, L. Botta, T. Semeraro, C. Mugnaini, A. Ligresti, E. Palazzo, et al. J. Med. Chem. 2008, 51, 5075.
| Crossref | GoogleScholarGoogle Scholar | 18680276PubMed |
[160] K. J. Valenzano, L. Tafesse, G. Lee, J. E. Harrison, J. M. Boulet, S. L. Gottshall, et al. Neuropharmacology 2005, 48, 658.
| Crossref | GoogleScholarGoogle Scholar | 15814101PubMed |
[161] C. Mugnaini, A. Brizzi, A. Ligresti, M. Allara, S. Lamponi, F. Vacondio, et al. J. Med. Chem. 2016, 59, 1052.
| Crossref | GoogleScholarGoogle Scholar | 26756097PubMed |
[162] C. Mugnaini, A. Rabbito, A. Brizzi, N. Palombi, S. Petrosino, R. Verde, et al. Eur. J. Med. Chem. 2019, 161, 239.
| Crossref | GoogleScholarGoogle Scholar | 30359820PubMed |
[163] Y. Shi, Y. H. Duan, Y. Y. Ji, Z. L. Wang, Y. R. Wu, H. Gunosewoyo, et al. J. Med. Chem. 2017, 60, 7067.
| Crossref | GoogleScholarGoogle Scholar | 28726401PubMed |
[164] S. M. Usama, E. R. Thapaliya, M. P. Luciano, M. J. Schnermann, Curr. Opin. Chem. Biol. 2021, 63, 38.
| Crossref | GoogleScholarGoogle Scholar | 33684856PubMed |
[165] H. J. Chen, C. Y. Chew, E. H. Chang, Y. W. Tu, L. Y. Wei, B. H. Wu, et al. J. Am. Chem. Soc. 2018, 140, 5224.
| Crossref | GoogleScholarGoogle Scholar | 29587477PubMed |
[166] Y. Ni, J. Wu, Org. Biomol. Chem. 2014, 12, 3774.
| Crossref | GoogleScholarGoogle Scholar | 24781214PubMed |