

Nitrogen- and Sulfur-Containing Energetic Compounds. 64 Years of Fascinating Chemistry*
Curt Wentrup AA School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Qld 4072, Australia. Email: wentrup@uq.edu.au
Australian Journal of Chemistry 72(8) 585-594 https://doi.org/10.1071/CH19263
Submitted: 10 June 2019 Accepted: 11 July 2019 Published: 6 August 2019
Abstract
This essay details the author’s work with high-energy molecules based on sulfur or nitrogen, or both, which started with amateur rocket propellants like zinc dust and sulfur followed by experiments with the highly sensitive compounds nitrogen trichloride and fulminating gold. Research on the inorganic and organic fulminates and the isomeric cyanates led to detailed investigations of reactive intermediates generated by flash vacuum pyrolysis or photolysis, in particular nitrenes and carbenes derived from azides, diazo compounds, triazoles, and tetrazoles and characterized in low temperature matrices.
Introduction
Not so long ago, it was common for professors to perform demonstration experiments in undergraduate classes, especially in the big first-year course on general chemistry. The idea was very much along the lines of the old German dictum ‘Die Chemie ist was knallt und stinkt’ (chemistry is that which bangs and stinks). Lecture theatres had fume hoods and long benches for experiments fitted with water, gas, etc. Experiments with sound effects were always very popular for teachers and students alike. The preparation and explosion of nitrogen triiodide, NI3, used to be a popular teaching experiment in undergraduate and even high school lectures. This compound is easily made by pouring ammonia onto iodine. Once dry, it explodes on the slightest touch. In junior high school I was lucky to have very knowledgeable teachers keen on hands-on demonstrations. It was not uncommon for kids to start experiments at home as soon as we learned something new in school.
I set up my first laboratory in the fall of 1955, soon after my 13th birthday. Because we had physics before chemistry, my first experiments were in electricity, electromagnetism, and the like in the tradition of H. C. Ørsted. Then, after some chemistry lectures, I began experimenting with chemistry. Growing up on a farm, I was fortunate to have access to some test tubes and glass vessels of various kinds left behind by the milk controller, who came around regularly on his motorbike to check the purity, fat content, and bacterial content of the different kinds of milk. I made good use of his discarded glassware in my experiments.
My first home-made rockets were propelled with ‘rocket candy’, which is a mixture of saltpeter and sugar. It is still widely used in amateur rockets, often using sorbitol as the fuel, which have reached altitudes of over 12 km.[1] By the time Sputnik I was launched in 1957, I was using a mixture of zinc dust and sulfur. Unbeknown to me, kids in the USA were doing the same.[2]
The reaction between zinc and sulfur produces ZnS as a greenish-yellow smoke precipitating as a yellowish powder,[3,4] and, depending on the ratio and efficiency of mixing, chunks of highly triboluminescent crystalline conglomerates of yellow zinc sulfide were sometimes formed. The combination of Zn and S is quite spectacular and highly exothermic (Fig. 1), but the specific impulse is rather low. It can be increased by adding extra oxidizers such as the then readily available saltpeter (potassium nitrate) or ammonium nitrate (a fertilizer) or sodium chlorate (a weed killer) and fuels such as aluminium powder or carbon.

Nitrogen Trichloride
A fascinating teach-yourself-chemistry book[5] from the municipal library taught us that we could make nitrogen trichloride, NCl3, by neutralizing the ammonium nitrate fertilizer and adding chlorine, which we did, well aware that it is a very dangerous experiment. The French physicist and chemist Pierre Louis Dulong first prepared NCl3 in 1811, and lost three fingers and an eye in the process.[6] A year later Sir Humphry Davy rediscovered NCl3 and was temporarily blinded in an explosion: ‘a violent flash of light was perceived, with a sharp report; the tube and glass were broken into small fragments, and I received a severe wound in the transparent cornea of the eye, which has produced a considerable inflammation … and obliges me to make this communication by amanuensis. The experiment proves what extreme caution is necessary in operating on this substance, for the quantity I used was scarcely as large as a grain of mustard seed’.[7,8]
The accident allegedly motivated him to hire Michael Faraday (Fig. 2) as an assistant in 1813. Subsequently, the two were both injured in another NCl3 explosion.[8] The nitrogen halides are sensitive to the touch and dangerously explosive. Because NCl3 is so easily made from chlorine and ammonia, nobody should play around with mixing cleaning products and/or pool chemicals.

|
Having liberated the ammonia, the synthesis of NCl3 starts with the formation of chloramine, NH2Cl.
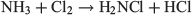
followed by
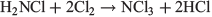
NH2Cl is formed in small quantities in swimming pools by reactions of chlorine with urea, dead skin cells, and some pool chemicals. It is more irritating to the eyes than the pool chlorine itself. NH2Cl has been and still is used as a disinfectant in municipal water works. This compound too has to be handled with care, as it is explosive in the neat state above −40°C.[9] Chloramine is an electrophilic aminating reagent, used for example in the synthesis of unsymmetrical dimethylhydrazine,

which has been used as a liquid rocket fuel by several nations together with liquid oxygen or N2O4 as the oxidant.[9]
Fulminating Gold
From the Bergsøe book[5] I also learned to make fulminating gold by dissolving gold in aqua regia and precipitating the salt by basification with ammonia. If the aqua regia is made from nitric acid and ammonium chloride, there are enough ammonium ions to generate NH3 on basifying with e.g. K2CO3 or Na2CO3. Otherwise, add an ammonium salt and then basify with a carbonate. The black precipitate thus formed is filtered off, washed with alcohol and ether, and allowed to dry in the air at a very low heat. Once dry, touching it with an iron instrument or heating causes it to explode with a very loud bang and giving off purple fumes of gold (Fig. 3).[10]
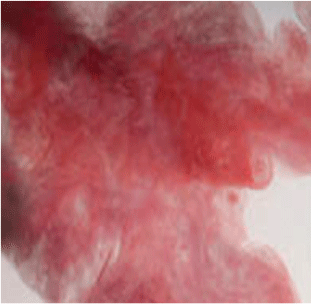
Fulminating gold (aurum fulminans) is not a fulminate but a polymeric condensation product of Au(NH3)43+. The counter ions can be Cl−, HO−, and O2− in various proportions depending on the method of preparation and isolation.[12] It was prepared by the German alchemist Sebald Schwaertzer[13,14] and described in a hand-written manuscript allegedly handed to the prince-elector (Kurfürst) of Saxony in 1583–1585, but it was only published in 1718.[15] There is a competing claim to priority by Basil Valentin,[16] in manuscripts allegedly written in the 15th century, but it is widely believed that the author was in fact Johan Thölde, who published the manuscripts no earlier than 1590.[17,18] However, Schwaertzer referred to ‘Bas. Val.’ in his manuscript, so the question arises whether Schwaertzer copied Valentin, or Valentin copied Schwaertzer. At any rate, Schwaertzer’s manuscript was copied by a Herr Tutschky, the Princely Saxonian Stall-Sekretarius, and there would have been plenty of time for further editing before the publication in 1718. It is noteworthy that, while they described very similar experiments, there is an important difference: Schwaertzer first precipitated his gold calx from aqua regia with copper vitriol (sulfate), then redissolved it in acid and precipitated it again with oleo Tartari (essentially K2CO3). The copper sulfate is unnecessary; all that is needed is that the solution contains ammonium ions and a base is added to neutralize the acid, so that the fulminating gold can precipitate. Valentin did not use the copper sulfate, nor did Croll, Hartmann, and Glauber (see below), who repeated the experiment later. It is particularly noticeable that Valentin wrote in typical alchemical, allegorical fashion, which is deliberately made very difficult for the uninitiated to understand, but Schwaertzer wrote in reasonably straightforward German.
Kunckel von Löwenstern (1630–1703)[19] worked more than 100 years after Schwaertzer, but he was also in Dresden for a period, and he reports having seen Schwaertzer’s original manuscript. Although Kunckel was an alchemist believing in the transmutation of metals, he was also an outstanding chemist, describing inter alia the first preparation of mercury fulminate (more of this below). He was highly supportive of Schwaertzer,[20] but clearly annoyed with Valentin’s difficult-to-understand style.[21] While the Schwaertzer–Valentin priority question remains open, and putting their claims to making gold by transmutation aside, there is no doubt that Schwaertzer reported important new chemistry, sanctioned by Kunckel, and his preparation of fulminating gold certainly works, although it is not the simplest procedure.
Oswald Croll[22] and Johannes Hartmann[23] were professors of medicine and chymiatry, respectively, in Marburg, and both published posthumous books on medicinal chemistry, including preparations of fulminating gold essentially based on Valentin’s prescription. The reason the iatrochemists were so interested in aurum fulminans is that it was used to make aurum potabile (drinkable gold), thought to be a medicinal panacea. Croll had found that the aurum fulminans is rendered unexplosive my mixing with sulfur, and Hartmann used this as his prescription for aurum potabile.[22] To this mixture add a menstruum of spiritus urinae (largely ammonium carbonate) and ethanol (spiritus vini) and heat it up to make a blood-red tincture. Distilling off the menstruum at low heat leaves a dark-red gold oil (probably largely Au2Cl6),[24] ‘which is the English aurum potabile according to Franz [Francis] Anthony’.[25] It was meant to be good for all illnesses and purified the body by inducing sweating.
Hartmann was the first chemistry professor in the world, and he also set up the first university chemistry teaching laboratory in the world, where students could learn his many preparations hands on. Among the many house rules for his laboratory, the students had to give the director (Hartmann) an oath of submission, loyalty, and diligence, coats and swords were under no circumstances allowed in the laboratories, the students were not to drink, sleep, shout, or fight, and if they behaved otherwise, they would be punished with a fine or banned from the community.
Johann Rudolf Glauber, the German–Dutch apothecary of Glauber Salt (Na2SO4) fame, used the deposition of the beautiful red-purple fumes generated in explosions of aurum fulminans (cf. Figs 3 and 4) for gold plating by means of an apparatus with two compartments connected with a glass pipe.[26] The fulminating gold (2–4 grain ~100–200 mg) was placed in the first compartment, and the object to be gilded in the second. Heating caused the fulminating gold to explode, and the explosion forced the purple fumes to stream through the connector and deposit on the object in the second compartment. The process was apparently efficient enough to be useful. He may have been able to use any gold deposited on the walls in the preparation of his gold tincture, which he described as the noblest medicine to strengthen the heart and purify the blood.
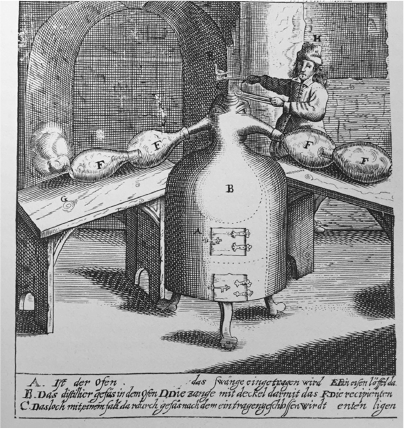
Furthermore, Glauber confirmed Croll’s observation that fulminating gold is made less dangerous by mixing with other compounds. This fact was reportedly used extensively by goldsmiths and enamellers in the 17th century. Painting with a mixture of aurum fulminans and flux and then firing the object produced the lustrous red-purple to coppery colouration (Böttger luster) due to finely divided gold.[18]
It was not only alchemists who played with fulminating gold. In the early 1800s the famous Jöns Jakob Berzelius of Sweden, the father of chemical formulae much like we use them today, was handling a quantity of as much as 9 g of the material, when it exploded, blinding him for months, and embedded numerous glass splinters in his hand.[27]
Diazotization and Diazo Compounds
At the age of 16, I discovered the wonderful and complex world of organic chemistry as an apprentice at Kemisk Værk Køge (now Sun Chemical), first in the preparation of blue and green phthalocyanine pigments, but more importantly diazotization, azo coupling, and the synthesis of red, orange, yellow, and gold-green azo dyes; for example, pigment yellow 12 (Scheme 1).

|
That gave me a valuable introduction to diazo chemistry, which would come in handy in my later work on cyanates, diazo compounds, and azides.[28]
Diazotization was invented by Peter Griess in 1858 (see further below). Griess was a farmer’s son from the German state of Hesse and studied at the University of Marburg. As was common in former centuries, students tended to be a rowdy lot given to drinking and public nuisance generally, for which reason universities regularly had their own prisons (cf. Hartmann’s rules above). Peter Griess was interned in the Karzer at least three times for unruly behaviour and was even banned from Marburg for a year.[29] He came back, and forced to earn a living he got a job in the Oehler factory. This changed him completely, he became hard-working and conscientious, and in 1857 he returned to the University to work under Professor Hermann Kolbe. Kolbe in turn helped him to get a position with A. W. Hoffmann at the Royal Institute (Imperial College), London. In Hoffmann’s laboratory, and later in the Alsopp & Sons brewery, Griess prepared and isolated many thermally highly unstable, explosive diazonium salts.[30] Moreover, he discovered azo coupling and prepared numerous azo dyes, and his work was of profound importance for the burgeoning dyestuff industry.[29]
Already in Kolbe’s laboratory, Griess had discovered the formation of diazo compounds by diazotization of aromatic amines, first picramic acid (2-amino-3,5-dinitrophenol) with N2O3 in ethanol,[31] which yielded the highly explosive diazo oxide (Scheme 2).
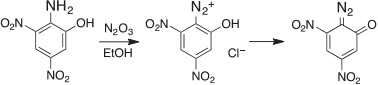
|
In my research group in Brisbane we have used the analogous benzene- and pyridine-diazo oxides in syntheses of (aza)fulven-6-ones (Scheme 3).[32]
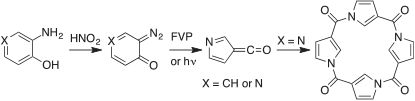
|
We have investigated numerous aryl- and heteroaryldiazo compounds and the rearrangements of the carbenes formed from them[28,33] by flash vacuum pyrolysis (FVP)[34] or photolysis.[28,35] Thus, FVP of the sodium salts of benzaldehyde and tropone tosylhydrazones produced heptafulvalene arising from the dimerization of cycloheptatetraene (CHT) (Scheme 4).[36,37]

|
Later, the triplet cycloheptatrienylidene (T) was observed by electron spin resonance (ESR) spectroscopy in the matrix photolysis of diazotropone, itself generated by FVP of the tosylhydrazone salt.[38] The phenylcarbene–cycloheptatrienylidene/cycloheptatetraene/fulvenallene rearrangement (Scheme 5) has been the subject of detailed theoretical studies.[39,40]

|
Cyanates, Isocyanates, Fulminates
During my undergraduate research at the University of Copenhagen I worked on the synthesis and spectroscopic and chemical properties of the newly discovered cyanates ROCN.[41] While the isocyanates, RNCO, had long been known, they had long been regarded as ‘cyanates’; for example, PhNCO was sold by E. Merck, Darmstadt, as ‘Phenylcyanat’ in the early 20th century. The ‘real’ alkyl cyanates had only just been discovered in the 1960s. This is because they are generally unstable, isomerizing to isocyanates or undergoing a facile elimination reaction to an alkene and HNCO.[42] Furthermore, the OCN function is a good leaving group, so that, in the presence of a nucleophile, the SN2 or SN1 substitution products are usually formed.[43]
The interest in the isomeric cyanates, isocyanates, and nitrile oxides RCNO goes back to the realisation by Liebig and Wöhler in 1827 that silver fulminate AgCNO and silver cyanate AgNCO have the same elemental composition, which contributed to Berzelius’ formulation of the concept of isomerism.[44,45] Examples cited by Berzelius were cyanic and fulminic acids, the two stannic acids, tartaric and racemic acids, and ammonium cyanate and urea. Fulminic acid has the structure HCNO,[46] i.e. it is formonitrile oxide. Cyanic acid, HOCN, rearranges very easily to isocyanic acid, HNCO, for example due to wall collisions when it is generated in FVP experiments, but it has been characterized by matrix IR and gas phase microwave spectroscopies.[47] Isofulminic acid, HONC, was elusive until a few years ago, when it was characterized by pulsed microwave spectroscopy.[48]
The dangerously explosive, shock sensitive mercury and silver fulminates were prepared by Kunckel von Löwenstern,[19] and more controversially by Cornelius Drebbel (1572–1633). In Kunckel’s procedure, to a mixture of silver and mercury in strong nitric acid was added spiritus vini, and the mixture was allowed to stand in warm horse manure till the next day, when it exploded with an enormous bang. Most likely, a mixture of mercury fulminate and the more sensitive silver fuminate had formed.
The Englishman Edward Howard and the Italian Luigi Brugnatelli prepared and isolated the pure mercury and silver fulminates, Hg(CNO)2 and AgCNO, in 1800–1805.[49–51] Howard isolated mercury fulminate as a grey powder by treating red mercuric oxide with nitric acid and ethanol and discovered that it exploded violently by addition of sulfuric acid. He was severely injured in the process.
I repeated Howard’s procedure as a teenager but luckily did not try pouring sulfuric acid on it! The preparation works fine.
It is interesting to note that Liebig,[52] on treating silver fulminate with ammonia, obtained another, violently explosive compound, which is the fulminating silver of Kunckel and Berthollet (argentum fulminans).[19,53] This compound is not a fulminate but a nitride, Ag3N, which, similar to NI3, is too sensitive to be handled.
Hg(CNO)2 has been used widely as a detonator, or blasting cap, to initiate the explosion of dynamite. Dynamite, which is used in blasting, was of course invented by Alfred Nobel and consists of nitroglycerin absorbed in diatomaceous earth (German Kieselguhr) to make it less shock sensitive. Nitroglycerin is very easy to prepare by nitration of glycerol, but it is too dangerous to be of any use; it may blow up if dropped on the floor. It was first synthesized by Ascanio Sobrero in 1847.[54] Dynamite is safer but requires a detonator.
In my research group in Marburg we developed a new and safe syntheses of fulminic acid and the fulminates, XONC, by thermal decomposition of 4-oximinoisoxazol-5(4H)-ones or their salts (Scheme 6).[55]
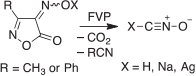
|
Fulminic acid is formed in virtually quantitative yield. It may be that the sought-after isofulminic acid, HONC, is formed first in this reaction, but it is not detectable under our reaction conditions. The barrier for isomerization HONC → HCNO is estimated to be only 105 kJ mol−1.[56] The fulminic acid undergoes partial isomerization to isocyanic acid, HNCO, at high temperature. Therefore, to obtain a high yield of fulminic acid, it is important to control the temperature of the FVP apparatus. We were able to prepare silver and sodium fulminates using the same method (Scheme 6).
The sulfur analogue, thiofulminic acid, HCNS, is much less stable than fulminic acid. While nitrile sulfides could be observed directly (Scheme 7), we were not able to detect HCNS in the analogous FVP reactions of oxathiazolone derivatives, but its existence as a stable, neutral species was established by neutralization–reionization mass spectrometry (Scheme 7).[57]
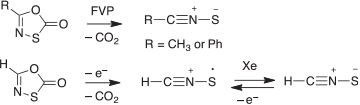
|
Meanwhile, HCNS and HSNC have also been characterized by microwave and matrix IR spectroscopies.[58]
Numerous chemists were trying to synthesize the organic fulminates (or isofulminates or fulminic acid O-esters) RONC since the 1800s; our method using FVP or photolysis of isoxazolones (Scheme 8) is so far the only successful one,[56,59] but the fulminates are extremely unstable, the calculated barriers to isomerization to cyanates are low, of the order of 100 kJ mol−1, and only small amounts can be detected in the FVP and photolysis products. Furthermore, in the phenyl case, a homolysis reaction eventually producing phenol and HCN is a major obstacle under FVP conditions.

|
In Copenhagen, to obtain the alkyl cyanates we first prepared xanthogenacetic acid esters according to Zeise (Fig. 5), who had discovered the xanthogenates, RO-CSSM, which are obtained by addition of alkoholates to CS2.[60] Zeise had also discovered the mercaptans (thiols) and did extensive experimentation with them.[61] The foul smells of the compounds clinging to his clothes did not make him popular with the other tenants where he lived or indeed with institutions in Copenhagen like the Royal Theatre, which denied him entry. One particularly nasty compound nicknamed the cat urine compound was 4-mercapto-4-methylpentan-2-one. Zeise made several other important discoveries, including the alkene-platinum π-complexes[62] now being investigated for applications in cancer chemotherapy.[63]

The alkali metal xanthogenates react with chloroacetic acid to form xanthogenacetic acids, and these are converted with hydrazine into xanthogen hydrazides,[64] which on diazotization cyclize to 5-alkoxythiatriazoles (Scheme 9). These compounds are very labile thermally, decomposing at or near room temperature to ROCN, N2, and ‘S’.[41,42,65]
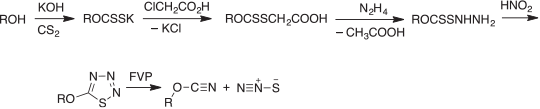
|
However, the sulfur formed by decomposition of the thiatriazoles is not atomic S1. It is isolated as S8 but, as I discovered much later, the highly unstable, linear molecule, dinitrogen sulfide (nitrous sulfide), N2S is an intermediate in this reaction (Scheme 9). It decomposes by dimerization to N2SSN2, which then dissociates into 2N2 + S2. Nonetheless, N2S is readily isolable from the FVP reaction at liquid nitrogen temperature. It was characterised by low temperature IR spectroscopy, and in the gas phase by mass and photoelectron spectroscopies.[66]
The photolysis of thiatriazoles leads to nitrile sulfides, RCNS, most probably by rearrangement of the initially formed thiazirines (Scheme 10).[66,67] Smaller amounts of isothiocyanates, RNCS, are also often formed in both photolysis and pyrolysis of 5-arylthiatriazoles,[66,67] probably by alternate rearrangement of the same thiazirine intermediates. The thiazirines can be regarded as the stable forms of the unknown singlet thioacylnitrenes. There are a few examples of observation of thiazirines by UV and IR spectroscopy.[68]
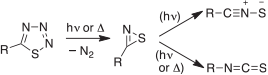
|
Similarly, singlet acylnitrenes exist in a partially ring-closed form, which can be considered a hybrid between a closed-shell singlet nitrene (CSS) and an oxazirine (Scheme 11).[28b] The triplet nitrene (T) is true nitrene. On photolysis of acyl azides, the singlet acylnitrenes rearrange to isocyanates (the Curtius rearrangement).[69]
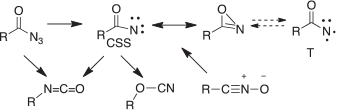
|
The acylnitrene/oxazirine hybrid was first put forward in 2000 as an intermediate in the rearrangement of nitrile oxides RCNO to isocyanates RNCO (Scheme 11), which takes place both thermally and photochemically.[70]
In the case of FVP of 5-alkoxy- and 5-aryloxythiatriazoles, we did not observe any rearrangement to alkoxy- or aryloxy isothiocyanates, RO-NCS, in the thermal decompositions. However, we were able to successfully generate and spectroscopically characterize alkoxy isothiocyanates by FVP of the silver salts of N-alkoxydithiocarbamic acids (Scheme 12).[71] The silver salt of N-methoxydithiocarbamic acid had been prepared previously by Traube, who recorded a sharp, pungent odour of the products of thermal decomposition, reminiscent of isothiocyanates.[72]
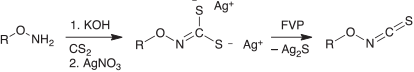
|
We have investigated the interconversions of acyl fulminates, cyanates, isocyanates and their thio derivatives, as well as imidoyl iso(thio)cyanates and (thio)acyl carbodiimides (Scheme 13)[73] and reviewed the preparations and rearrangements of heteroatom-substituted cyanates, isocyanates, and nitrile oxides and their thio derivatives.[74]

|
During my work on cyanates in 1966, a surprising observation was the formation of a peak at m/z 91 (C6H5N•+) in the mass spectrum of phenyl cyanate, PhOCN, and this became the base peak in the mass spectrum of phenyl isocyanate, PhNCO.[41a] I wondered what could be the structure of this ion, formally the phenylnitrene ion, Ph-N•+.
Azides
At the ANU, and later at the universities of Lausanne, Marburg, and Queensland, I was able to undertake a long series of investigations of the FVP of aryl azides,[75,76] and in particular to discover the ring contraction and ring expansion of phenylnitrene 2 to cyanocyclopentadiene 3 and azacycloheptatetraene 6 (Scheme 14).[77]
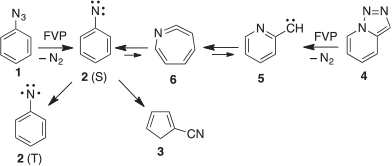
|
As we now know, these same processes are energetically very favourable in the radical cations in the mass spectrometer and therefore responsible for the generation of the strong m/z 91 (C6H5N•+) signal on electron impact of phenyl azide 1, triazolopyridne 4, phenyl isocyanate, and several other precursors.[78]
Phenyl azide (‘diazobenzolimide’) was first prepared by Peter Griess via the diazotization of aniline, which yields benzenediazonium bromide C6H5N2+Br−. With excess bromine this forms a ‘dibromide’, C6H5N2+Br3−, which reacts with excess ammonia to yield C6H5N3 + 3HBr (Fig. 6). Griess noted that the azide must be distilled under vacuum and detonates if exposed to higher heat.[30,79] Nowadays, aryl azides are more commonly prepared by addition of sodium azide to a diazonium salt solution, or by diazotization of arylhydrazines.

We have generated arylnitrenes from many azides, triazoloazines, and tetrazolylazines by FVP, whereby the carbene–nitrene rearrangement (5 → 6 → 2, Scheme 14) is a highly efficient source of nitrenes.[28b,75,76] Interestingly, the sulfur-nitrogen compound phenylsulfinylamine also undergoes FVP-induced cleavage to phenylnitrene, as evidenced by the formation of cyanocyclopentadiene and aniline in modest yields, together with the triplet state of SO, which was characterized by microwave spectroscopy (Scheme 15).[80] SO disproportionates rapidly to SO2 and S2O.
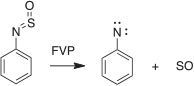
|
Although highly energetic, aromatic azides are relatively safe to handle and in many cases commercially available, i.e. they do not blow up, unless you are careless, e.g. by trying to distil them at atmospheric pressure. In spite of pyrolyzing numerous azides in literally thousands of FVP experiments at temperatures up to 1000°C, we have never had an accident. Most azides are easily evaporated in high vacuum, and heating them to up to 100°C in the solid or liquid state under vacuum has been unproblematic. However, it must be pointed out that gem-diazides are dangerously explosive (not to mention tri- and tetra-azides). For example, the azidotetrazoles and gem-diazides used to generate aryl isocyanides and the previously unknown sulfonyl isocyanides (Scheme 16)[81] are highly sensitive and violently explosive.
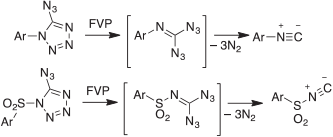
|
We took the necessary safety precautions and had no accidents. Nonetheless, this type of work should only be done on a very small scale.
Conclusion
Work with high-energy materials containing nitrogen and/or sulfur progressed from simple amateur rocket propellants to compounds like nitrogen trichloride, fulminating gold, and fulminates and their isomers the cyanates (organic and inorganic), and finally detailed investigations of reactive intermediates generated by flash vacuum pyrolysis and/or matrix photolysis, especially the nitrenes and carbenes generated from azides, diazo compounds, triazoles, and tetrazoles.
Conflicts of Interest
The author declares no conflicts of interest.
Acknowledgements
I would like to acknowledge my school teachers in Store Heddinge, who enthused me for chemistry at an early age, and to Professors K. A. Jensen (Copenhagen), W. D. Crow (ANU, Canberra), and H. Dahn (Lausanne) who encouraged and made it possible for me to undertake independent research. I am grateful to the many students and postdoctoral associates who since 1969 have helped keeping my research exciting and energetic in Switzerland (Lausanne), Germany (Marburg), and Australia (Queensland); only a few of them could be mentioned in the references. I am fortunate to be able to continue my research on reactive intermediates through fruitful collaborations with researchers in inter alia France, Germany, Japan, and China.
References
[1] Newshub, Auckland, 17 November 2013. Available at: https://www.newshub.co.nz/nznews/sugarfuelled-rocket-breaks-world-record-2013111717 (accessed 7 June 2019).[2] H. H. Hickam, The Rocket Boys 1998 (Fourth Estate: London).
[3] K. A. Boudreaux. Available at: https://www.angelo.edu/faculty/kboudrea/demos/zinc_sulfur/zinc_sulfur.htm (accessed 24 May 2019).
[4] For a description of the Zn-S reaction, see, for example, pp. 53–54 in B. Z. Shakhashiri, Chemical Demonstrations: A Handbook for Teachers of Chemistry, Volume 1 1983 (The University of Wisconsin Press: Madison, WI).
[5] P. Bergsøe, Kemi på en Anden Måde, Rosenkilde og Bagger, Copenhagen 1942.
[6] W. E. Hale, Science 1888, 11, 273.
[7] H. Davy, Phil Trans. Roy. Soc. 1813, 103, 1.
| Crossref | GoogleScholarGoogle Scholar |
[8] J. M. Thomas, Michael Faraday and The Royal Institution (The Genius of Man and Place) 1991 (CRC Press, Taylor and Francis Group: Boca Raton, FL).
[9] Chloramine: https://en.wikipedia.org/wiki/Chloramine (accessed 24 May 2019).
[10] Review on fulminating gold: C. Wentrup, Angew. Chem. Int. Ed. 2019, in press.
| Crossref | GoogleScholarGoogle Scholar |
[11] Das Gold, das kracht. Available at: https://sciencev1.orf.at/news/154577.html (accessed 1 June 2019).
[12] G. Steinhauser, J. Evers, S. Jakob, T. Klapötke, G. Oehlinger, Gold Bull. 2008, 41, 305.
| Crossref | GoogleScholarGoogle Scholar |
[13] Sebald(t) Schw(a)ertzer biography: Sächsische Biographie, Institut für Sächsische Geschichte und Volkskunde e.V.: http://saebi.isgv.de/biografie/Sebalt_Schwertzer_(1552-1598).
[14] See pp. 350–351 in: J. Ferguson, Bibliotheca Chemica, Vol. 2 1954 (Academic and Bibliographical Publishing Ltd: London).
[15] S. Schwaertzer, Chrysopoeia Schwaertzeriana, 1585, pp. 84–86. Reprinted by Samuel Heil, Hamburg, 1718, under the title ‘Chrysopoeia Schwaertzeriana. Das ist: Sebaldi Schwaertzers, ehemaligen berühmten Churfürstl. Sächsischen Artisten und würcklichen Adepti, Manuscripta, Von der Wahrhafften Bereitung des Philosophischen Steins: Wie selbige vor diesem mit seiner eigenen Hand entworffen, und bey dem Chur-Fürstl. Sächsischen Hause in Originali verwahrlich aufbehalten worden, Nebst dem rechten zu solchen Manuscriptis gehörigen Schlüssel; Auch unterschiedlichen Abrissen der darzu dienliche Ofen, Aus einer unverfälschten durch viele Mühe und Unkosten erlangten Copia nunmehro jederman vor Augen geleget, und mit einigen nützlichen Anhängen von verschiedenen curieusen Processen vermehret’. Available at: https://reader.digitale-sammlungen.de/de/fs1/object/display/bsb10253002_00014.html (accessed 1 April 2019).
[16] Basil Valentine biographies: (a) https://en.wikipedia.org/wiki/Basil_Valentine. (b) J. Ferguson, Bibliotheca Chemica, Vol. 1 1954 (Academic and Bibliographical Publishing Ltd: London), pp. 81–82.
[17] See pp. 150–169 in: L. M. Principe, The Secrets of Alchemy 2013 (The University of Chicago Press: Chicago, IL).
[18] N. Zumbyliadis, Bull. Hist. Chem. 2014, 39, 7.
[19] Johann Kunckel von Löwenstern, Collegium Physico-Chymicum Experimentale Oder Laboratorium Chymicum, 1738; facsimile reprint, Kessinger Publishing, Whitefish, MT, USA, 2010. The manuscript was written about 1700 and published posthumously.
[20] Support of Schwaertzer: Laboratorium Chymicum pp. 586, 592–606.
[21] Critique of Valentin: Laboratorium Chymicum pp. 391, 454.
[22] O. Croll, Basilicum Chymicum, by Ioan. Hartmanno, edited by Iohanne Michaelis and Georg Everhardo Hartmanno, Samuel Chouët, Geneva 1658. Available at: http://dfg-viewer.de/show/?tx_dlf%5Bpage%5D=73&tx_dlf%5Bdouble%5D=1&tx_dlf%5Bid%5D=http%3A%2F%2Fdigital.ub.uni-duesseldorf.de%2Foai%2F%3Fverb%3DGetRecord%26metadataPrefix%3Dmets%26identifier%3D1882248&cHash=caf88ba0b16b27f4b080cbb4381ee127 (accessed 2 April 2019).
[23] J. Hartmann, Praxis Chymiatriae, posthumous edition by Johanna Michaelis and Georgio Everharto Hartmanno, Gotfred Grossi, Leipzig 1633.
[24] See pp. 150–169 in: L. M. Principe, The Secrets of Alchemy 2013 (The University of Chicago Press: Chicago).
[25] F. Anthonie, The Apologia or Defence of a Verity Hertofore Published Concerning a Medicine called Avrum Potabile...As An Universal Medicine 1616 (Iohn Legart: London). Available at: https://archive.org/details/b30339479/page/114 (accessed 10 May 2019).
[26] Johannis Rudolphi Glauberi, Operum Chymicorum, Bücher und Schriften so viel deren von ihme bisshero an Tag gegeben worden. Jetzo von neuem Traktaten vermehret…Frankfurt 1659. https://play.google.com/books/reader?id=23Y5veS13eQC&hl=en&pg=GBS.PA2-IA6 (accessed 1 April 2019).
[27] M. Speter, Nitrocellulose 1930, 1, 128.
[28] (a) Rearrangements and interconversions of carbenes and nitrenes: C. Wentrup, Top. Curr. Chem. 1976, 62, 173.
| Crossref | GoogleScholarGoogle Scholar | 941142PubMed |
(b) Carbenes and nitrenes minireview: C. Wentrup, Angew. Chem. Int. Ed. 2018, 57, 11508.
| Crossref | GoogleScholarGoogle Scholar |
[29] R. Wizinger-Aust, Angew. Chem. 1958, 70, 199.
| Crossref | GoogleScholarGoogle Scholar |
[30] P. Griess, Proc. R. Soc. Lond. 1864, 13, 275.
[31] P. Griess, Liebigs Ann. Chem. Pharm. 1858, 106, 123.
| Crossref | GoogleScholarGoogle Scholar |
[32] (a) G. G. Qiao, M. W. Wong, C. Wentrup, J. Org. Chem. 1996, 61, 8125.
| Crossref | GoogleScholarGoogle Scholar | 11667800PubMed |
(b) R. Koch, R. J. Blanch, C. Wentrup, J. Org. Chem. 2014, 79, 6978.
| Crossref | GoogleScholarGoogle Scholar |
[33] C. Wentrup, Acc. Chem. Res. 2011, 44, 393.
| Crossref | GoogleScholarGoogle Scholar | 21452850PubMed |
[34] Flash vacuum pyrolysis: C. Wentrup, Angew. Chem. Int. Ed. 2017, 56, 14808.
| Crossref | GoogleScholarGoogle Scholar |
[35] For a review on heteroarylcarbenes, see: R. S. Sheridan, Chem. Rev. 2013, 113, 7179.
| Crossref | GoogleScholarGoogle Scholar | 23799781PubMed |
[36] C. Wentrup, C. Wilczek, Helv. Chim. Acta 1970, 53, 1459.
| Crossref | GoogleScholarGoogle Scholar |
[37] (a) See also: R. C. Joines, A. B. Turner, W. M. Jones, J. Am. Chem. Soc. 1969, 91, 7754.
| Crossref | GoogleScholarGoogle Scholar |
(b) P. Schissel, M. E. Kent, D. J. McAdoo, E. Hedaya, J. Am. Chem. Soc. 1970, 92, 2147.
| Crossref | GoogleScholarGoogle Scholar |
[38] M. Kuzaj, H. Lüerssen, C. Wentrup, Angew. Chem. Int. Ed. Engl. 1986, 25, 480.
| Crossref | GoogleScholarGoogle Scholar |
[39] (a) S. Matzinger, T. Bally, E. V. Patterson, R. J. McMahon, J. Am. Chem. Soc. 1996, 118, 1535.
| Crossref | GoogleScholarGoogle Scholar |
(b) P. R. Schreiner, W. L. Karney, P. R. Schleyer, W. T. Borden, T. P. Hamilton, H. F. Schaefer, J. Org. Chem. 1996, 61, 7030.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. W. Wong, C. Wentrup, J. Org. Chem. 1996, 61, 7022.
| Crossref | GoogleScholarGoogle Scholar |
[40] D. Kvaskoff, H. Lüerssen, P. Bednarek, C. Wentrup, J. Am. Chem. Soc. 2014, 136, 15203.
| Crossref | GoogleScholarGoogle Scholar | 25322946PubMed |
[41] (a) K. A. Jensen, A. Holm, C. Wentrup, J. Møller, Acta Chem. Scand. 1966, 20, 2107.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. Holm, C. Wentrup, Acta Chem. Scand. 1966, 20, 2123.
| Crossref | GoogleScholarGoogle Scholar |
[42] C. Wentrup, Acta Chem. Scand. 1966, 20, 2128.
| Crossref | GoogleScholarGoogle Scholar |
[43] K. A. Jensen, M. Due, A. Holm, C. Wentrup, Acta Chem. Scand. 1966, 20, 2091.
| Crossref | GoogleScholarGoogle Scholar |
[44] (a) R. Winderlich, J. Chem. Educ. 1948, 25, 500.
| Crossref | GoogleScholarGoogle Scholar |
(b) See p. 45 in: J. J. Berzelius, Årsberättelse om framstegen i Physik och Chemie, Stockholm 1831.
[45] See pp. 257–260 in: J. R. Partington, A History of Chemistry, Vol. 4 1964 (Macmillan: New York, NY).
[46] (a) W. Beck, Eur. J. Inorg. Chem. 2003, 4275.
| Crossref | GoogleScholarGoogle Scholar |
(b) W. Beck, G. Fischer, P. Swoboda, Z. Anorg. Allg. Chem. 2017, 643, 353.
| Crossref | GoogleScholarGoogle Scholar |
[47] (a) V. E. Bondybey, J. H. English, C. W. Mathews, R. J. Contolini, J. Mol. Spectrosc. 1982, 92, 431.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. Brünken, C. A. Gottlieb, M. C. McCarthy, P. Thaddeus, Astrophys. J. 2009, 697, 880.
| Crossref | GoogleScholarGoogle Scholar |
[48] M. Mladeovic, M. Lewerenz, M. C. McCarthy, P. Thaddeus, J. Chem. Phys. 2009, 131, 1743908.
[49] E. Howard, Philos. Trans. R. Soc. Lond. 1800, 90, 204.
| Crossref | GoogleScholarGoogle Scholar |
[50] (a) L. Brugnatelli, Phil. Mag. 1803, 16, 185–186 and 258.
(b) L. Brugnatelli, Nicholson’s J. 1804, VII, 285.
[51] F. Kurzer, J. Chem. Educ. 2000, 77, 851.
| Crossref | GoogleScholarGoogle Scholar |
[52] J. Liebig, Büchners Repertorium der Pharmacie 1822, 12, 412.
[53] C. L. Bertholet, Procédé Pour rendre la Chaux d’Argent fulminante; Observations sur la Physique, sur l’Histoire Naturelle et sur les Arts, 1788, 32, 474–476.
[54] A. Sobrero, Compt. Rend. Hebd. Séanc. Acad. Sci. 1847, 24, 247.
[55] C. Wentrup, B. Gerecht, H. Briehl, Angew. Chem. Int. Ed. Engl. 1979, 18, 467.
| Crossref | GoogleScholarGoogle Scholar |
[56] T. Pasinszki, M. Krebsz, Curr. Org. Chem. 2011, 15, 1688.
| Crossref | GoogleScholarGoogle Scholar |
[57] P. Kambouris, M. Plisnier, R. Flammang, J. K. Terlouw, C. Wentrup, Tetrahedron Lett. 1991, 32, 1487.
| Crossref | GoogleScholarGoogle Scholar |
[58] (a) B. A. McGuire, M.-A. Martin-Drumel, S. Thorwirth, S. Brünken, V. Lattanzi, J. L. Neill, S. Spezzano, Z. Yu, D. P. Zaleski, A. J. Remijan, B. H. Patee, M. C. McCarthy, Phys. Chem. Chem. Phys. 2016, 18, 22693.
| Crossref | GoogleScholarGoogle Scholar | 27478937PubMed |
(b) T. Pasinszki, M. Krebs, G. Bazso, G. Tarczay, Chem. – Eur. J. 2009, 15, 6100.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Wierzejewska, Z. Mielke, Chem. Phys. Lett. 2001, 349, 227.
| Crossref | GoogleScholarGoogle Scholar |
[59] C. Wentrup, B. Gerecht, D. Laqua, H. Briehl, H.-W. Winter, H. P. Reisenauer, M. Winnewisser, J. Org. Chem. 1981, 46, 1046.; Erratum J. Org. Chem. 1981, 46, 5459
| Crossref | GoogleScholarGoogle Scholar |
[60] (a) W. C. Zeise, Ann. Phys. Chem. (Poggendorff) 1834, 32, 305.
| Crossref | GoogleScholarGoogle Scholar |
(b) W. C. Zeise, Ann. Phys. Chem. (Poggendorff) 1835, 35, 487.
| Crossref | GoogleScholarGoogle Scholar |
[61] W. C. Zeise, Ann. Chim. Phys. 1834, 56, 87.
[62] (a) W. C. Zeise, Pogg. Ann 1827, 9, 632.
(b) W. C. Zeise, Magazin für Pharmacie 1830, 36, 105.
(c) W. C. Zeise, Pogg. Ann 1831, 21, 497.
(d) W. C. Zeise, Schweiggers J. Chem. Phys. 1831, 62, 393.
(e) W. C. Zeise, Schweiggers J. Chem. Phys. 1831, 63, 121.
[63] A. Weninger, D. Baecker, V. Obermoser, D. Egger, K. Wurst, R. Gust, Int. J. Mol. Sci. 2018, 19, 1612.
| Crossref | GoogleScholarGoogle Scholar |
[64] K. A. Jensen, U. Anthoni, A. Holm, Acta Chem. Scand. 1969, 23, 1916.
| Crossref | GoogleScholarGoogle Scholar |
[65] K. A. Jensen, A. Holm, B. Thorkilsen, Acta Chem. Scand. 1964, 18, 825.
| Crossref | GoogleScholarGoogle Scholar |
[66] C. Wentrup, P. Kambouris, Chem. Rev. 1991, 91, 363.
| Crossref | GoogleScholarGoogle Scholar |
[67] A. Holm, Adv. Heterocycl. Chem. 1976, 20, 145.
| Crossref | GoogleScholarGoogle Scholar |
[68] (a) A. Tajti, L. A. Mück, Á. L. Farkas, M. Krebsz, T. Pasinszki, G. Tarczay, P. G. Szalay, J. Mol. Spectrosc. 2015, 310, 8.
| Crossref | GoogleScholarGoogle Scholar |
(b) T. Vörös, G. G. Lajgút, G. Magyarfalvi, G. Tarczay, J. Phys. Chem. A 2018, 122, 1034.
| Crossref | GoogleScholarGoogle Scholar |
[69] C. Wentrup, H. Bornemann, Eur. J. Org. Chem. 2005, 4521.
| Crossref | GoogleScholarGoogle Scholar |
[70] M. Barbieux-Flammang, S. Vandevoorde, R. Flammang, M. W. Wong, H. Bibas, C. H. Kennard, C. Wentrup, J. Chem. Soc., Perkin Trans. 2 2000, 473.
| Crossref | GoogleScholarGoogle Scholar |
[71] A. T. Bech, R. Flammang, C. Th. Pedersen, M. W. Wong, C. Wentrup, J. Chem. Soc., Perkin Trans. 2 1999, 1869.
| Crossref | GoogleScholarGoogle Scholar |
[72] W. Traube, H. Ohlendorf, H. Zander, Ber. Dtsch. Chem. Ges. 1920, 53, 1477.
| Crossref | GoogleScholarGoogle Scholar |
[73] R. Koch, C. Wentrup, J. Org. Chem. 2013, 78, 1802.
| Crossref | GoogleScholarGoogle Scholar | 22954414PubMed |
[74] C. Wentrup, J. J. Finnerty, R. Koch, Curr. Org. Chem. 2011, 15, 1745.
| Crossref | GoogleScholarGoogle Scholar |
[75] FVP of azides, triazoles and tetrazols: C. Wentrup, Chem. Rev. 2017, 117, 4562.
| Crossref | GoogleScholarGoogle Scholar | 28234461PubMed |
[76] C. Wentrup, in Nitrenes and Nitrenium Ions (Eds D. E. Falvey, A. D. Gudmundsdottir) 2013, Ch. 8, pp. 273–315 (John Wiley & Sons: Hoboken, NJ).
[77] (a) W. D. Crow, C. Wentrup, Tetrahedron Lett. 1967, 8, 4379.
| Crossref | GoogleScholarGoogle Scholar |
(b) W. D. Crow, C. Wentrup, Tetrahedron Lett. 1968, 9, 6149.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. Kvaskoff, H. Lüerssen, P. Bednarek, C. Wentrup, J. Am. Chem. Soc. 2014, 136, 15203.
| Crossref | GoogleScholarGoogle Scholar |
[78] D. Bégué, A. Dargelos, C. Braybrook, C. Wentrup, J. Phys. Chem. A 2018, 122, 8490.
| Crossref | GoogleScholarGoogle Scholar | 30296825PubMed |
[79] P. Griess, Justus Liebigs Ann. Chem. 1865, 135, 121.
| Crossref | GoogleScholarGoogle Scholar |
[80] (a) S. Saito, C. Wentrup, Helv. Chim. Acta 1971, 54, 273.
| Crossref | GoogleScholarGoogle Scholar |
(b) C. Wentrup, Tetrahedron 1971, 27, 1027.
| Crossref | GoogleScholarGoogle Scholar |
[81] H. Briehl, C. Wentrup, ChemRxiv 2017, 20, preprint.
| Crossref | GoogleScholarGoogle Scholar |
* The author was awarded the 2018 Leighton Medal of the RACI in recognition of service to chemistry.